Epidemiology, Cytogenetic, and Molecular Aspects
Disorder associated with a fragile site or a partial break on the long arm of the X chromosome at the q27.3 region. In 1991 the mutated gene that causes fragile X syndrome was identified and sequenced. It is called the Fragile X Mental Retardation 1 gene or FMR1 and is located at Xq27.3. A trinucleotide repetitive sequence (CGG) was found within FMR1, such that normal individuals have approximately 6 to 50 repeats, carrier individuals have 50 to 200 repeats (premutation), and individuals who are affected by fragile X syndrome have >200 CGG repeats (full mutation).
The full mutation is associated with methylation of the FMR1 gene, which turns off transcription and translation of the FMR1 gene into protein. It is the absence of the FMR1 gene protein (FMRP) that causes fragile X syndrome.
The clinical picture of fragile X syndrome includes mental retardation, often with autistic-like characteristics such as poor eye contact, hand-flapping, and hand-biting. Physical features are also associated with this syndrome, including macroorchidism or large testicles, a long narrow face, and large or prominent ears. Hyperextensibility of the finger joints, flat feet, and mitral valve prolapse are also commonly seen clinically. Males who are affected with fragile X syndrome are usually retarded, although the spectrum of involvement ranges from learning disabilities with a normal IQ to profound retardation. All males with the full mutation are clinically affected by the syndrome, that is, they usually demonstrate mental retardation and associated physical features. Approximately one-half of females with the full mutation are cognitively impaired with a borderline IQ or with mental retardation. Individuals who carry the premutation (50 to 200 CGG repeats) are usually unaffected intellectually.
Because this disorder is carried on the X chromosome, it is inherited in an X-linked fashion, that is, it is passed on from mother to son and cannot be passed on from father to son. The full mutation occurs only when the mutation is passed on by a mother who has the premutation or a full mutation. Carrier males with the premutation will pass on the premutation only to all of their daughters but to none of their sons, who receive the Y chromosome. The overall incidence of individuals affected with this syndrome in the general population is approximately 1 in 2,000.
The fragile X chromosome (Fig. 1) was first visualized in 1969 in a family with four retarded males over three generations. Work in Australia led to the association of the fragile X chromosome with the physical phenotype in the mid-1970s, however, individuals were rarely diagnosed with this disorder during the 1970s in the United States. This was because the demonstration of the fragile X chromosome is dependent on the use of a specific type of tissue culture media that was described by G. Sutherland in the late 1970s.


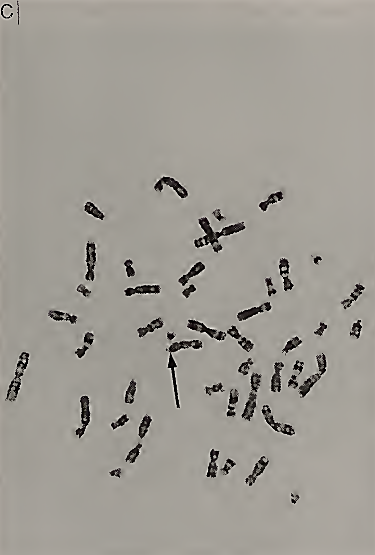
FIGURE I. (A) A scanning electron micrograph of chromosomes in metaphase with arrow pointing to the fragile site on the X chromosome. (B) Closeup of two fragile X chromosomes with the lowest arrow pointing to the fragile site at Xq27.3. [Reprinted with permission from C. J. Harrison et al. (1983). The fragile X: A scanning electron microscope study./. Med. Genet. 20, 280-285.] (C) Metaphase spread viewed from light microscopy. The arrow is pointing to the fragile site at q27.3 on the X chromosome
The cytogenetic testing done on a peripheral blood sample requires that the cells be grown in tissue culture media that is deficient in folic acid and/or thymidine. Expression of the fragile X chromosome can also be induced by inhibitors of folic acid, such as methotrexate, or by inhibitors of thymidylate synthetase, such as 2-deoxy-5-fluorouridine (FUdR). Under these conditions, the fragile X chromosome will be seen in 1 to 50% of the cells analyzed. Although all the cells carry a mutation at the fragile X site, the visualization of the fragile site at Xq27.3 is not seen in every cell for unknown reasons.
Usually only individuals with the full mutation demonstrate the fragile site. Individuals with the premutation are usually negative on cytogenetic studies but they are still at high risk to have children with fragile X syndrome and the full mutation (Fig. 2). An analysis of the FMR1 gene can be done with DNA testing of a blood sample.
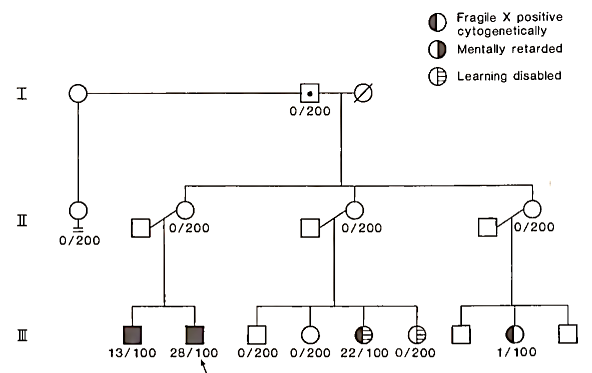
FIGURE 2. A fragile X pedigree. Cytogenetic results (number of fragile X positive lymphocytes/total number of lymphocytes evaluated) are noted below each patient. Generation I has a nonpenetrant male (grandfather of affected males) who has been documented to carry the fragile X gene by DNA studies
DNA testing is less expensive and more accurate than cytogenetic testing for fragile X syndrome or carrier status. However, DNA testing for FMR1 mutation does not include testing of other genes or structural defects throughout the genome, which is done in cytogenetic testing. Therefore, in the workup of mental retardation or autism of unknown etiology, both DNA and chromosome testing are usually carried out, whereas in the workup of a known fragile X family, only the DNA testing is necessary to identify carriers with the premutation and individuals more affected with the full mutation.
Date added: 2022-12-11; views: 793;