Porphyrins and Bile Pigments
Porphyrins are heterocyclic compounds composed of four pyrrole rings linked to each other by four methene bridge carbon atoms (Fig. 1). These cyclic compounds are extremely stable, having been found in petroleum shale oil and in fossilized excrements millions of years old.
The basic structures of porphyrins are found in the heme molecules of hemoglobin, myoglobin, cytochromes, catalases, and the like, in the chlorophylls and bacteriochlorophylls of photosynthetic organisms, in siroheme and coenzyme F430, and in the corrin ring of vitamin B12 (Fig. 2).
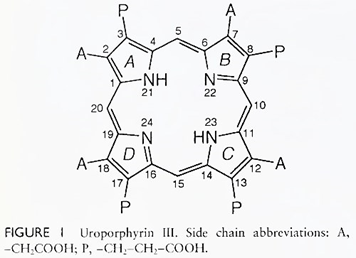
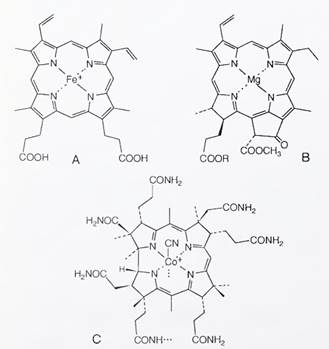
FIGURE 2. Structures of (A) heme, (B) chlorophyll, and (C) the corrin ring of vitamin B12
The porphyrin ring of heme is coordinated to iron, whereas chlorophyll contains magnesium, coenzyme F430 contains nickel, and the corrin ring is linked to cobalt. The heme proteins are concerned with oxygen transport (hemoglobin), oxygen storage (myoglobin), oxidation (cytochromes and peroxidases), and nitrogen fixation (leghemoglobin). Whereas heme is concerned primarily with the release of available energy, the role of chlorophyll is photosynthesis, by which solar energy builds up reducing and oxidizing potentials, so as to store chemical energy.
As discussed in Section I, the biosynthesis of porphyrins involves the formation of a “parent” porphyrin structure having an acetic acid (A) and a propionic acid (P) side chain in each of the ß positions of each of the pyrrole rings. With an A and a P side chain in each of the pyrrole rings, one can construct four structural isomers.
The parent structures of all biological functioning porphyrins or porphyrin-derived compounds have the following distribution of side chains, starting from ring A (see Fig. 1): АР, AP, AP, PA; they are designated isomer III. In particular pathological or metabolic altered states, isomer I (AP, AP, AP, AP) is formed as well.
The unique structures of the side chains eventually found in the functioning porphyrin (e.g., protoporphyrin of heme) arise by subsequent alteration of the A and P side chains of the parent isomer III: for example, the methyl side chains of protoporphyrin are derived by decarboxylation of the four acetic acid (A) side chains, and the two vinyl side chains (rings A and B) are derived by decarboxylation and oxidation of the propionic acid (P) side chains (Figs. 1 and 3).
Biosynthesis of porphyrins. The first committed step in the synthesis of the porphyrin structure, and thereby in the synthesis of heme, chlorophyll, and the corrin ring, is the formation of δ-aminolevulinic acid (ALA). In humans as well as other members of the animal kingdom, and also in photosynthetic bacteria, ALA is enzymatically synthesized from the simple amino acid glycine and from succinic acid with the participation of two coenzymes—coenzyme A and pyridoxal phosphate— which are derivatives of two essential human dietary compounds: panthothenic acid and vitamin B6, respectively (see Fig. 3).
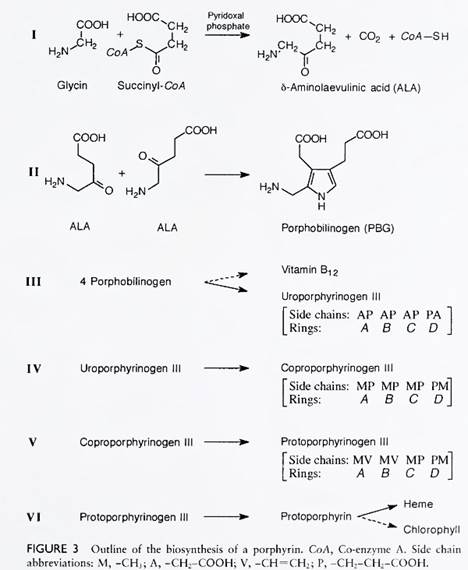
It was recently found that vertebrates have two separate genes that encode for the enzyme that catalyzes this reaction (i.e., δ-aminolevulinate synthase). The erythroid gene is expressed exclusively in the red blood cell, whereas the hepatic form of the enzyme is probably expressed ubiquitously. It is of further interest that plants, algae, and other microorganisms (e.g., Cyanobacteria, Chromatium and Methauobacterinm) synthesize ALA from glutamic acid, an amino acid.
Subsequently, two molecules of ALA are enzymatically condensed to form a pyrrole derivative, porphobilinogen (PBG). The enzyme catalyzing this reaction (i.e., PBG synthase) is a zinc-containing enzyme markedly inhibited by the presence of lead. Four molecules of PBG are then condensed enzymatically with the participation of two enzymes (PBG deaminase and uroporphyrinogen III cosynthase) to form uroporphyrinogen III (reduced uroporphyrin III). (In the absence of cosynthase, the nonbiological functioning isomer I is formed.)
The A side chains of uroporphyrinogen III are enzymatically decarboxylated to methyl groups to form coproporphyrinogen III and subsequently the P side chains of rings A and В are decarboxylated and dehydrogenated to vinyl groups to form protoporphyrinogen. The latter is oxidized to protoporphyrin, which is enzymatically converted to heme by the addition of iron.
It is worth noting that the first step (in Fig. 3) and last three enzymatic steps occur in the mitochondria, whereas the intermediate enzymes are found in the cytosol. The rate-limiting step in the synthesis of porphyrins appears to be the synthesis of ALA, partially regulated by the concentration of heme. Chlorophyll and the corrin ring of vitamin B12 are synthesized from intermediates in the outlined scheme (see Fig. 3).
Porphyrias. There are approximately seven well-defined human inborn errors in the metabolic process concerned with the synthesis of porphyrins. The genetic inheritance is usually autosomal dominant, except for congenital erythropoietic porphyria and ALA dehydratase deficiency porphyria, which are autosomal recessive disorders. Two of these disorders are briefly described here.
A. Congenital Erythropoietic Porphyria. Congenital erythropoietic porphyria is a rare disease in which the homozygote is characterized by an increase of porphyrins in the blood, urine, and feces. Since the abnormality lies in the deficiency of uroporphyrinogen III cosynthase, the excretion is that of uroporphyrin I and coproporphyrin I. The accumulation of the porphyrins in the tissues, bones, and teeth also causes the individual to be photosensitive, exposure to sunlight causing blisters that readily become infected. The bones and the teeth of individuals with this disorder are reddish. A similar disorder has been found in cattle and in the fox squirrel.
B. Acute Intermittent Porphyria. Acute intermittent porphyria is an autosomal dominant disorder in which the metabolic defect is in the deficiency of the enzyme porphobilinogen deaminase. Approximately 50% of the normal enzymatic activity is found in the subjects’ tissues.
Most of the subjects who inherit this genetic deficiency do not exhibit any clinical symptoms or biochemical abnormalities. However, when exposed to some drugs (e.g., barbiturates or sulfonamides) or some stresses, they accumulate PBG and ALA and express clinical symptoms. Along with marked increases in PBG and ALA in the urine, the subjects can have acute abdominal pains, neurological symptoms, paralysis, muscle weakness, and some abnormal behavioral patterns.
It is of interest that, before being recognized as a metabolic disorder, many subjects, in the acute phase, have received appendectomies and even been assigned to mental institutions. The incidence of the disease is apparently more common in Scandinavia, South Africa, Great Britain, and Lapland. The occurrence in some in stances has been traced to the original individual carrying the defective gene. The injection of hemin (heme-Fe3+), which inhibits ALA synthesis, appears to lessen the symptoms occurring in the acute phase.
Date added: 2024-07-02; views: 639;