Cell Specificity and Susceptibility
In HD and ALS, the particular vulnerability of neurons to altered proteostasis could be the cause of the selective neurodegeneration observed in these diseases. Unlike neurons, which do not divide, dividing cells can dilute the misfolded protein through uneven division and the younger cells eventually no longer carry the misfolded or aggregated proteins. Neurons and potentially other specific cell types in the brain are particularly vulnerable to these toxic proteins because of their different ability to employ UPS, autophagy, or the chaperone network.
For example, the activity of UPS is lower in the striatal cells when compared to cortical cells. This would lead to a reduced ubiquitination and clearance of the mutant HTT in the striatum, causing cellular dysfunction and death. The protein chaperone network, which assists in folding proteins, has distinct expression levels in different regions of the brain (Margulis and Finkbeiner, 2014). By analogy similar changes in the proteostasis network may give clues to the selective neurodegeneration found in ALS.
Symptoms and Treatments of the Disease. The physical manifestations of both HD and ALS on an organismal level are profound, both diseases exhibit massive neuronal death, enough to be distinguished on autopsies of the deceased patients (Figure 2).
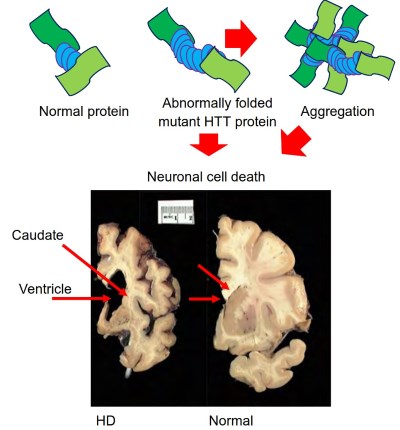
Figure 2. Huntington's disease protein, aggregation and pathology. The upper panel is a comparison of HTT protein with a normal CAG repeat size compared to a mutant HTT with an expanded CAG repeat size and the aggregation. Macroscopic image in which a slice of Huntington's brain (left) is put next to a slice from a normal control (right).
Note that the striatum (the caudate nucleus, putamen, and nucleus accumbens), on the left, is severely atrophied in the Huntington's brain. Note also that the cerebral cortex is atrophied in the Huntington's brain. Photo courtesy of the Harvard Brain Tissue Resource Center
This massive neuronal death disrupts the brain affecting the patient's ability to perform movements and in many cases cognitive functions. In HD the region with the highest incidence of neuronal death is the striatum, particularly the medium spiny neurons. Medium spiny neurons are GABAergic neurons, which have an inhibitory effect on the neurons they are in contact with, these neurons are essential in the coordinating movement (Tepper et al., 2008; Walker, 2007).
The progressive neurodegeneration leads to in chorea, a movement disorder associated with HD, in which the patient exhibits spastic, involuntary movements. Tetrabenazine, is currently one of USDA approved drug for treating chorea, it works by inhibiting dopamine receptors (Zheng et al., 2006; Huntington Study, 2006; Poon et al., 2010). Beyond the physical symptoms, HD patients experience cognitive decline, depressive episodes, mood swings, and obsessive behavior (Montoya et al., 2006; van Duijn et al., 2007).
The behavioral and cognitive changes increase as the disease progresses, treatment involves the standard psychiatric medications. The mutant HTT is expressed throughout the body tissues, leading to muscle atrophy, cardiac failure, impaired glucose tolerance, osteoporosis, and weight loss (van der Burg et al., 2009). These symptoms can be treated with a combination of physical therapy, diet changes, and medications. There are no HD specific drugs or treatments and there is no cure, the eventual outcome of the disease is death.
ALS progresses at a much more rapid rate than HD, usually resulting in death within 3-5 years of the first symptoms (Robberecht and Philips, 2013). In ALS the patient first notices difficulties moving an extremity, this progresses toward muscle atrophy, whole body movement difficulty, and eventually respiratory failure (Kiernan et al., 2011).
These symptoms are due to axonal retraction, death of the neuron, and denervation of the muscles, resulting in paralysis (de Carvalho et al., 2010). In a subpopulation of patients the prefrontal and temporal cortex are affected to a degree which results in dementia (Phukan et al., 2007). A subsection of the patient population does not experience any cognitive decline and is fully aware of their slow paralysis.
The progression and severity of symptoms is dependent on both the type of genetic mutation and the age at onset of symptoms, creating a very heterogeneous population of patients in symptoms and survival time (Kiernan et al., 2011; Hardiman et al., 2011). No cure exists and medications are mostly focused on reducing symptom severity, Riluzola has shown some promise in increasing survival time, if given in the early stages of the disease (Carlesi et al., 2011).
Date added: 2024-06-13; views: 558;