The Role of Monocytes and Lymphocytes
Monocytes that have become resident in the sub- endothelial space can undergo several histologic transitions. When exposed to macrophage colony-stimulating factor (M-CSF), the monocyte converts into a macrophage. Macrophages are one of the earliest histologic substrates of atherogenesis. Macrophage egress from the vessel wall can be inhibited by the neural guidance factor netrin-1 and VCAM-1; egress can be potentiated by lymphatic channels and high-density lipoprotein particles.
The fatty acids and phospholipids of low-density lipoprotein particles can undergo oxidation via such enzymes as myeloperoxidase, NADPH oxidase, and a variety of lipoxygenases (Fig. 2.4). These oxidized phospholipid species are complex and potentiate inflammation and oxidation (Fig. 2.5). In addition, in the setting of hyperglycemia, lipoproteins can undergo glycation.
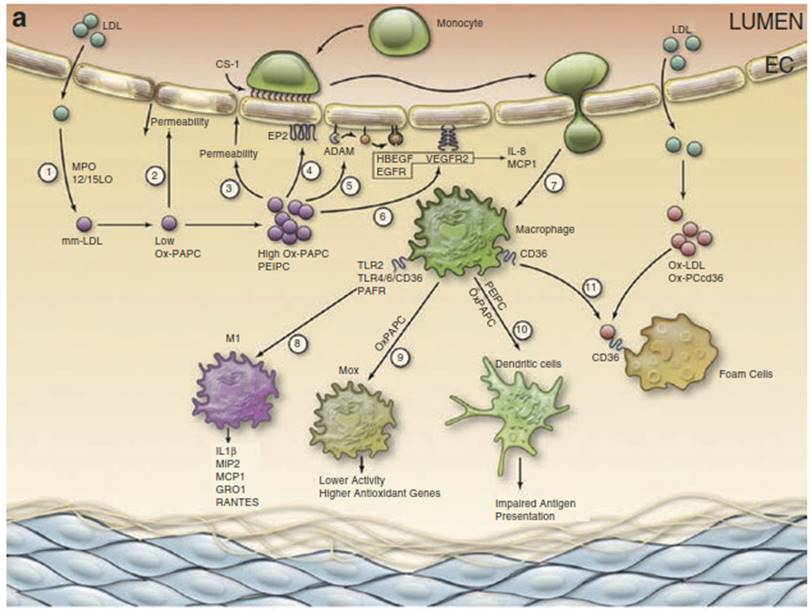
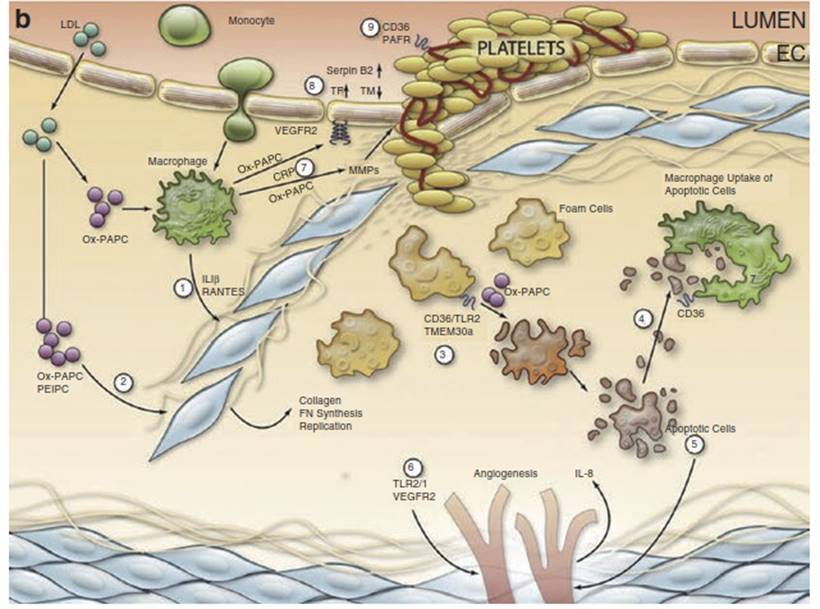
Fig. 2.4 Model of oxidized phosphatidylcholine- containing phospholipids (Ox-PL) regulation of atherosclerosis. (a) Early lesions: (1) LDL enters the vessel wall and is oxidized by myeloperoxidase (MPO) and 12/15 lipoxygenase (LO) to form modified LDL which contains oxidation products including oxidized 1-palmitoyl-2- arachidonyl-sn-glycero-3-phosphocholine (Ox-PAPC).
(2) Low doses of Ox-PAPC decrease the permeability of the endothelial cell (EC) monolayer by forming adherens junctions.
(3) Higher levels of Ox-PAPC cause a strong increase in monolayer permeability because of junction breakdown and stress fiber formation, resulting in increased entry of LDL into the vessel wall.
(4) Ox-PAPC binds to the E-type prostaglandin receptor (EP2) receptor, causing the deposition of connecting segment 1 (CS-1) fibronectin on the apical surface which binds monocytes.
(5) Ox-PAPC activates specific a disintegrin and metalloproteinases (ADAMs) to cause the release of active heparin-binding epidermal growth factor (HBEGF) and activation of epidermal growth factor receptor (EGFR), leading to interleukin (IL)-8 and monocyte chemotactic protein (MCP)-1 synthesis.
(6) Ox-PAPC also activates vascular endothelial growth factor receptor 2 (VEGFR2), leading to IL-8 and MCP-1 synthesis.
(7) These chemokines facilitate the entry of monocytes into the vessel wall.
(8) Oxidized phosphatidylcholine-containing phospholipids (Ox-PL) acting on Toll-like receptor (TLR)2, TLR4/6/cluster determinant 36 (CD36), and platelet-activating factor (PAF) receptor cause some monocytes to differentiate into M1 macrophages producing chemokines.
(9) Ox-PAPC causes differentiation of some macrophages into Mox, which have high levels of antioxidant enzymes and lower chemokine syntheses.
(10) Ox-PAPC causes the differentiation of some monocytes into dendritic cells with an impaired presentation of lipid antigens.
(11) Macrophages further oxidize LDL to form Ox-LDL. Ox-PCCD36 acting on CD36 in the presence of Ox-LDL causes foam cell formation.
(b) Advanced lesions: (1) In the presence of Ox-PAPC and PAF- like lipids, macrophages make IL-1 beta (IL-1P) and regulated upon activation, normal T-cell expressed, and secreted (RANTES).
(2) These chemokines and a direct effect of Ox-PAPC on smooth muscle celsl (SMC) cause the migration and proliferation and matrix production of SMC. These SMC cover the foam cells that accumulate under the endothelium.
(3) The interaction of Ox-PAPC with CD36/TLR2 and with unfolded protein response (UPR) activators and the interaction of PAF-like lipids with transmembrane protein 30A (TMEM30a) cause macrophage apoptosis.
(4) Oxidized phosphatidylserine-containing phospholipids (Ox-PS)/ PCCD36 in the apoptotic cell membrane bind to CD36 in macrophages, leading to macrophage uptake of the apoptotic cells.
(5) Some apoptotic fragments stimulate EC to make IL-8, an angiogenic cytokine.
(6) CEP activation of TLR2/TLR1 causing integrin activation and Ox-PAPC acting to increase VEGFA cause angiogenesis of adventitial vessels into the media and intima.
(7) C-reactive protein (CRP) and Ox-PAPC interacting with CD36 stimulate macrophage production of metalloproteinase. This weakens the plaque and can lead to plaque rupture.
(8) Ox-PAPC activation of VEGFR2 increases tissue factor synthesis in the endothelium. Ox-PAPC also causes increases in Serpin B2 and a decrease in thrombomodulin.
(9) Ox-PCCD36 acting on CD36 and PAF-like lipids acting on PAFR cause increased aggregability of platelets
Exposure to oxidatively modified or glycated low-density lipoprotein particles trapped by intercellular matrix proteins in the subendothelial space stimulates macrophages to upregulate the expression of a number of scavenger receptors on their surface. There are a large number of these scavenger receptors and include multiple types of scavenger receptor A (types I-III), CD36, lectin-like oxidized LDL receptor-1 (LOX-1), and scavenger receptor for phosphatidylserine and oxidized LDL (SR-PSOX), among others. LOX-1 is also expressed by endothelial cells and smooth muscle cells. In the setting of increased oxidized LDL (oxLDL) exposure, endothelial cells upregulate LOX-1; the uptake of oxLDL is toxic and potentiates endothelial dysfunction and adhesion molecule expression.
The oxidation of phospholipids in LDL particles generates the formation of oxidation specific epitopes recognized by scavenger receptors. These include oxidized sn-2 fatty acids that terminate in γ-hydroxy-α, β-unsaturated carbonyl groups or 1-palmitoyl-2-(5'-oxovaleroyl)-sn- glycero-3-phosphocholine (Fig. 2.5). Scavenger receptors promote the binding and uptake of atherogenic lipoproteins into the intracellular space of the macrophage. As more and more lipids are taken up, the macrophage develops lipid inclusion bodies and becomes a “foam cell.” Foam cells produce a variety of cytokines, matrix metalloproteinases, ROS, and tissue factors.
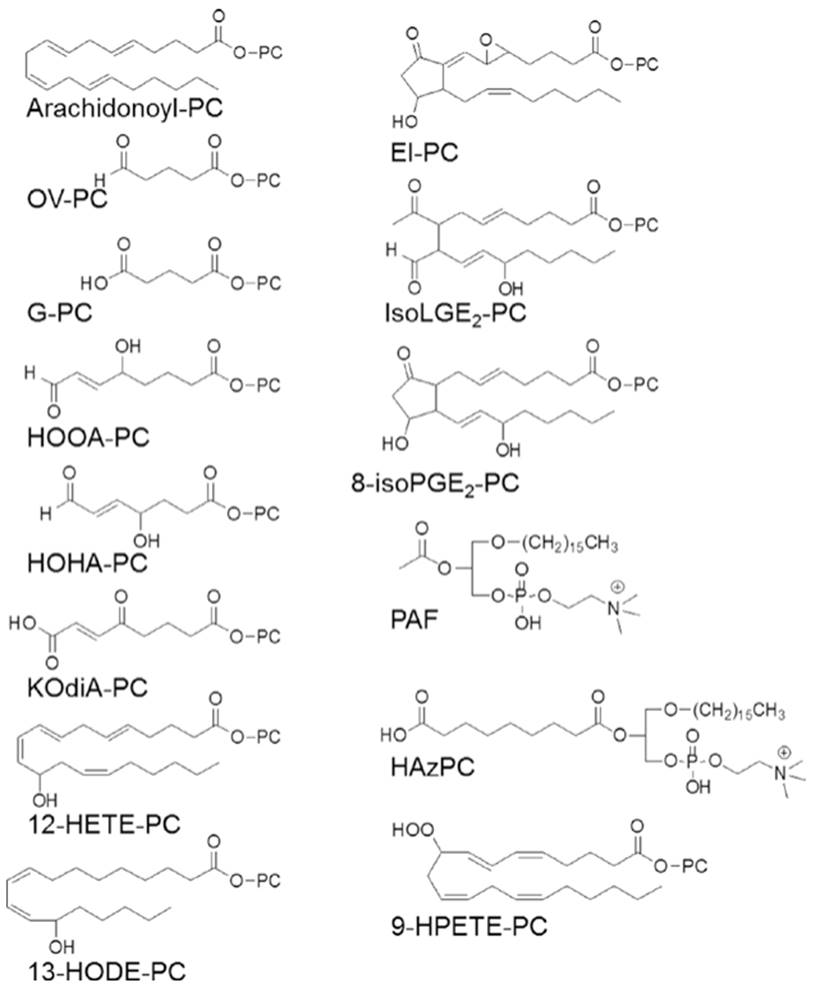
Fig. 2.5. Oxidized phosphatidylcholinecontaining phospholipids (OX-PL) lipids. PC, 1-acyl-2-lyso-sn-glycero-3phosphatidylcholine. Only the sn-2 position composition is shown for all Ox-PL except those forming an ether bond at the sn-1 position. Abbreviations: PAF platelet-activating factor, HAz-PC hexadecylazelaoyl PC, 13-HODE-PC 1-palmitoyl-2-(13(S)- hydroxy-(9Z,11E) octadeca-9,11-dienoyl)- sn-glycero-3- phosphocholine
Smooth muscle cells can undergo transformation into macrophages and, as they scavenge lipid and lipoprotein, can also form foam cells. Smooth muscle cell transformation occurs secondary to reduced expression of myocardin and the microRNAs miR143/145. Oxidized phospholipids can also stimulate the hyperphosphorylation of VE-cadherin, a critical protein for maintaining endothelial gap junctions. Gap junction function deteriorates as the VE-cadherin dissociates from the proteins p-catenin and paxillin.
Tissue factor is a procoagulant that promotes platelet aggregation on the surface of ruptured atheromatous plaques. The MMPs can destabilize atheromatous plaque by hydrolyzing the matrix proteins which reinforces its structural integrity (Fig. 2.6). As MMPs degrade extracellular matrix material, such degradation products as integrin-binding fibronectin, hyaluronan, and heparan sulfate can trigger immune and proinflammatory responses. Smooth muscle cells also produce MMPs as they break down the internal elastic lamina in order to access the intima. Ultimately, foam cells can coalesce to form fatty streaks. As fatty streaks increase in volume and more cellular debris accumulates, a frank atheromatous plaque evolves.
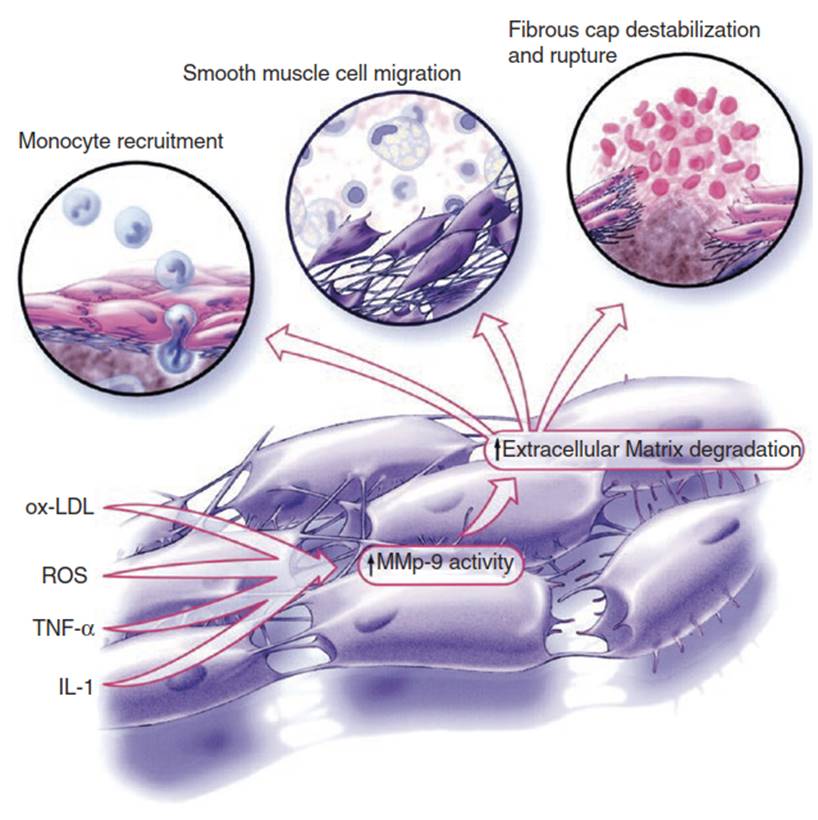
Fig. 2.6. MMP-9, from plaque progression to destabilization. MMP-9 degrades the basement membrane of the arterial wall to facilitate monocyte migration into the plaque and SMC migration to form the fibrous cap. Excessive MMP-9 activity eventually leads to the degradation of the fibrous cap, plaque instability, and plaque rupture. Abbreviations: IL-1 interleukin-1, ROS reactive oxygen species, TNF-α tumor necrosis factor alpha, MMP-9 matrix metalloproteinase-9, oxLDL oxidized low-density lipoprotein
Foam cells possess measures of self-defense. Macrophages are capable of effluxing excess intracellular lipids into the extracellular space. Intracellular cholesterol can be mobilized and exported onto HDL particles via scavenger receptor B-I (SR-BI), or two ATP-binding membrane cassette transport proteins termed ABCA1 and ABCG1. In addition, the macrophage can produce and secrete apoprotein E (apoE) which, when externalized, can bind to ABCA1 and drive cholesterol externalization, apoE lipidation, and lipoprotein biogenesis. If these defenses are overwhelmed by excess lipid trapped in the subendothelial space, then foam cell development progresses with increasing lipid inclusion body volume and resultant toxicity.
T lymphocytes and mast cells also participate in vascular inflammation and atherogenesis. T cells follow a gradient of chemoattractants (inducible protein-10, interferon-inducible T-cell α-chemoattractant, and monokine induced by interferon-γ) into the subendothelial space. These chemoattractants can bind to CXCR3, a chemokine receptor on the surface of T cells. When a T cell binds oxidatively modified LDL to an antigen receptor it can undergo differentiation into T helper cells, such as TH1 and TH2. TH1 cells potentiate inflammation by producing interleukin-1, interferon-γ, and tumor necrosis factor. TH2 cells produce anti-inflammatory cytokines, such as interleukins -4 and -10. TH1 cells predominate in atheromatous plaques and stimulate inflammation. Following antigen binding and presentation, T cells stimulate macrophage production of MMPs and cytokines.
Lymphocytes also infiltrate and become organized in the vascular adventitia. Adventitial aortic tertiary lymphoid organs (ATLOs) and T cell aggregates associate with more severe atherosclerotic plaques. An ATLO is composed of a nodular center composed of B lymphocytes and dendritic cells surrounded by T lymphocytes. B lymphocytes can be activated to produce antibodies after antigen presentation by dendritic cells, thereby mounting an immune response. There is significant communication between the endothelium and adventitia, and it is believed that ATLOs and organized T cell aggregates play a significant role in atherogenesis. The vasa vasora and small medial conduits mediate the transfer of immune cells, cytokines, and interleukins between the intima and adventitia (Fig. 2.7).

Fig. 2.7. Arterial adventitia and its role in atherogenesis. Lymphocytes, macrophages, dendritic, cells, and plasma B cells can be organized in the adventitia of arteries. Small medial conduits facilitate the passage of cytokines, chemokines, soluble antigens, and growth factors from the adventitia into the media. The vasa vasora can facilitate communication between cells of the adventitia and those of the intima, including endothelium. These communication patterns can promote atherogenesis by stimulating inflammatory and phagocytic cell recruitment, smooth muscle cell migration, and the mounting of an innate immune response
Activated mast cells contribute to atherogenesis and enter the subendothelial space in response to eotaxin exposure. Mast cells secrete two serine peptidases, tryptase and chymase. Chymase catalyzes the intravascular conversion of AI to AII, and tryptase activates MMPs. Both enzymes thus not only contribute to early events in atherogenesis but also can induce instability in established plaque. Mast cells also secrete histamine, which promotes increased vascular permeability.
Date added: 2025-02-17; views: 433;