Diseases Affecting Primarily Low-Density Lipoprotein
Familial Hypercholesterolemia. Introduction. Familial hypercholesterolemia (FH) is a disorder encompassing a group of genetic defects resulting in severely elevated serum cholesterol concentrations. It is a commonly inherited condition characterized by abnormal regulation of cholesterol metabolism causing severely elevated plasma levels of low-density lipoprotein cholesterol (LDL-C).
Although FH was specifically attributed to mutations of the LDL receptor (LDLR) in the past, the definition has been expanded to include gain-of-function mutations in proprotein convertase subtilisin kexin 9 genу (PCSK9), as well as mutations in the apolipoprotein (Apo) B gene (APOB) (Fig. 3.1) and the more recently described LDLR adaptor protein 1 gene (LDLRAP1). In the Fredrickson classification, most patients will fall under type IIa, with predominantly elevated LDL-C levels and normal triglycerides (TGs); other forms such as type IIb have been described less frequently.
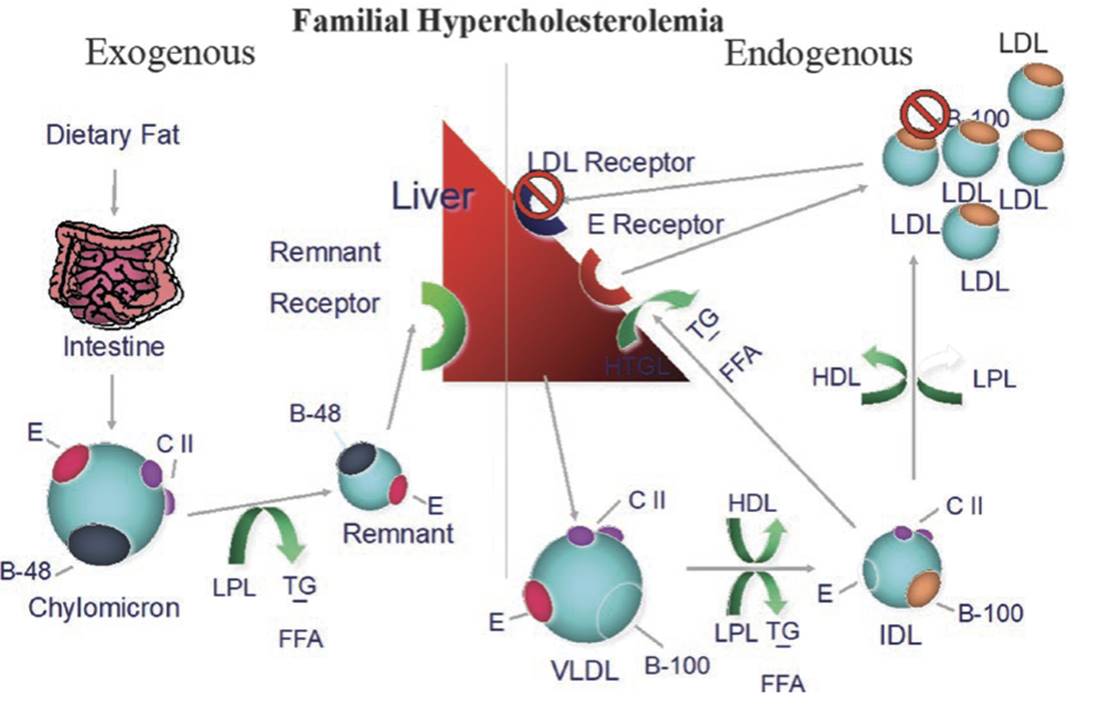
Fig. 3.1. Familial hypercholesterolemia is most commonly due to a mutation in the gene for the LDL receptor, causing receptor dysfunction. More rarely, a mutation in ApoB can cause the disorder due to poor binding affecting of LDL to the LDL receptor. A third etiology, a gain-of-function mutation in the gene for PCSK9 can lead to receptor destruction, which is extremely rare. All three etiologies lead to inability to clear LDL from the circulation and marked elevation of LDL levels
Prevalence. FH is the most common autosomal dominant genetic disorder, recognizing that non-dominant forms do exist, but are rare. The prevalence in the general population is between 1:200 and 1:500 depending on which criteria are used for the diagnosis. Certain populations, such as French Canadian, Dutch Afrikaner, Ashkenazi Jewish, and Lebanese Christian, have a prevalence of 1:100 or more. In the United States, an estimated 600,000-1000,000 individuals are affected. Among patients with premature coronary artery disease (CAD), the prevalence is much higher, reaching 5%, and increases as the age of the first cardiovascular (CV) event decreases.
Among diagnosed individuals, around 80% carry an identifiable mutation for specific genes. They comprise 85-90% affecting LDLR, 5-10% affecting APOB, less than 5% affecting PCSK9, and less than 1% affecting LDLRAP1 [15-22]. FH without an identifiable mutation comprises 20% of all cases. The majority is autosomal dominant in transmission, and most patients carry the heterozygous form, with LDL-C levels in the range of 350-550 mg/dL. The homozygous form is extremely rare, occurring in approximately 1 out of a million individuals. It is also much more severe, with LDL-C levels ranging between 650 and 1000 mg/dL.
All forms of FH are associated with a significantly elevated risk of premature CAD (around 20-fold in untreated cases), but in homozygous mutations that risk is exceedingly high. In general, “compound homozygous FH” (individuals carrying a different mutation on each allele) may have less risk than true homozygous FH where both alleles carry the same mutation. Studies suggest that FH is underdiagnosed in up to 80% of cases, and even confirmed cases are frequently undertreated. In the cases of homozygous children, a delay in treatment can be catastrophic; the therapeutic window for intervention may be missed before an individual develops their first CV event.
Genetics. LDLR: The LDLR is responsible for removing LDL-C from the plasma circulation. This process requires synthesis of adequate amounts of protein followed by appropriate transport to the Golgi apparatus, with subsequent expression on the cell surface. The LDLR protein needs to have the capacity to interact with LDL-C (through ApoB receptors), and internalize with the bound particles before being recycled back to the hepatocyte surface in order to maintain adequate LDL-C clearance.
The gene for LDLR resides on the short arm of chromosome 19. Any genetic defect affecting the quantity or functional properties of the LDLR protein can consequently lead to significantly increased levels of plasma LDL-C. Genetic defects may result from nonsense, missense, insertion, deletion, or splicing mutations. In total, there are more than 3000 recognized variants of the LDLR gene, with over 1600 pathogenic mutations identified to cause FH. Phenotypically, allele mutations, and their resultant protein abnormality, can be divided into 5 classes.
- Class I mutations, also known as “null mutations,” result in complete lack of LDLR synthesis.
- Class II mutations give rise to transport abnormalities with partially or completely retained protein within the endoplasmic reticulum.
- Class III mutations are associated with defective binding and consequent inability to interact with ApoB100 on the LDL surface.
- Class IV mutations do not permit proper endocytosis, interfering with LDL-LDLR complex internalization.
- Class V mutations produce proteins with defective recycling.
ApoB (Familial Defective APOB): The ApoB100 protein is normally expressed on the surface of LDL and serves as a ligand for LDLR. Pathogenic mutations of the APOB gene can result in a defective ApoB100 protein, interfering with LDL particle-receptor interaction, and effectively decreasing LDL-C clearance. The Arg3500Gln mutation is the most common cause of FH associated with defective ApoB and is frequently seen in northern European populations. Other pathogenic variants exist as well, and ApoB mutations are considered the second most common after LDLR, accounting for 5-10% of the cases. Typically, LDL particle levels increase by two- or three-fold, as opposed to more significant elevations seen in LDLR mutations.
PCSK9: PCSK9 is a serine protease, which gained much recognition in the early 2000s. It binds LDLR and forms an LDLR-PCSK9 complex that undergoes endocytosis followed by destruction within hepatocytes. This process does not allow for LDLR recycling and leads to diminished availability of active LDLR, thus causing increased levels of circulating LDL particles.
PCSK9 variants can be associated with loss-of- function or gain-of-function mutations, with the latter resulting in more active protein. Thus far, more than 30 variants of gain-of-function mutations have been identified, with associated FH as a consequence. On the other hand, loss of function is more commonly encountered in the general population with important clinical effects. Less active PCSK9 protein allows for more available LDLR, and subsequently more clearance of LDL particles. This implication has been utilized clinically with the advent of a new class of medication (PCSK9 inhibitors) that is currently used in the treatment of FH. Collectively, FH resulting from PCSK9 gain-of-function mutations are not very common and represent less than 5% of all documented FH cases in many studies.
LDLRAP1: Since the 2011 National Lipid Association Expert Panel Executive Summary, newer genetic mutations have been identified within the LDLRAP1 sequence that affect the LDL-LDLR interaction. LDLRAP1 protein mediates LDL-LDLR complex internalization by allowing for proper clathrin-coated endosome formation. The end result of dysfunctional endocytosis is limited clearance of LDL particles and increased plasma levels. This entity forms the fourth recognized defect to cause FH and differs by being an autosomal recessive disorder requiring the presence of two abnormal alleles before FH is manifested phenotypically. Overall, it accounts for less than 1% of all cases.
Date added: 2025-02-17; views: 505;