Role of Increased Oxidative Tone. Atheromatous Plaque
Myeloperoxidase, lipoprotein-associated phospholipase A2, xanthine oxidase, NADPH oxidase, cyclooxygenase, and 5'-lipoxygenase are all found in atheromatous plaque and promote ROS production and oxidative lipoprotein modification. The ROS include superoxide anion, hydroxyl radicals, peroxynitrite radicals, and hydrogen peroxide (Fig. 2.3).
Enzymes such as glutathione peroxidase, the thioredoxins, paraoxonase, and superoxide dismutase are responsible for metabolizing ROS to less reactive species. Deficiencies in anti-oxidative enzymes can be associated with increased atherogenesis. All the major cardiovascular risk factors (dyslipidemia, cigarette smoking, hypertension, diabetes mellitus) increase oxidative tone by upregulating the speciation of ROS.
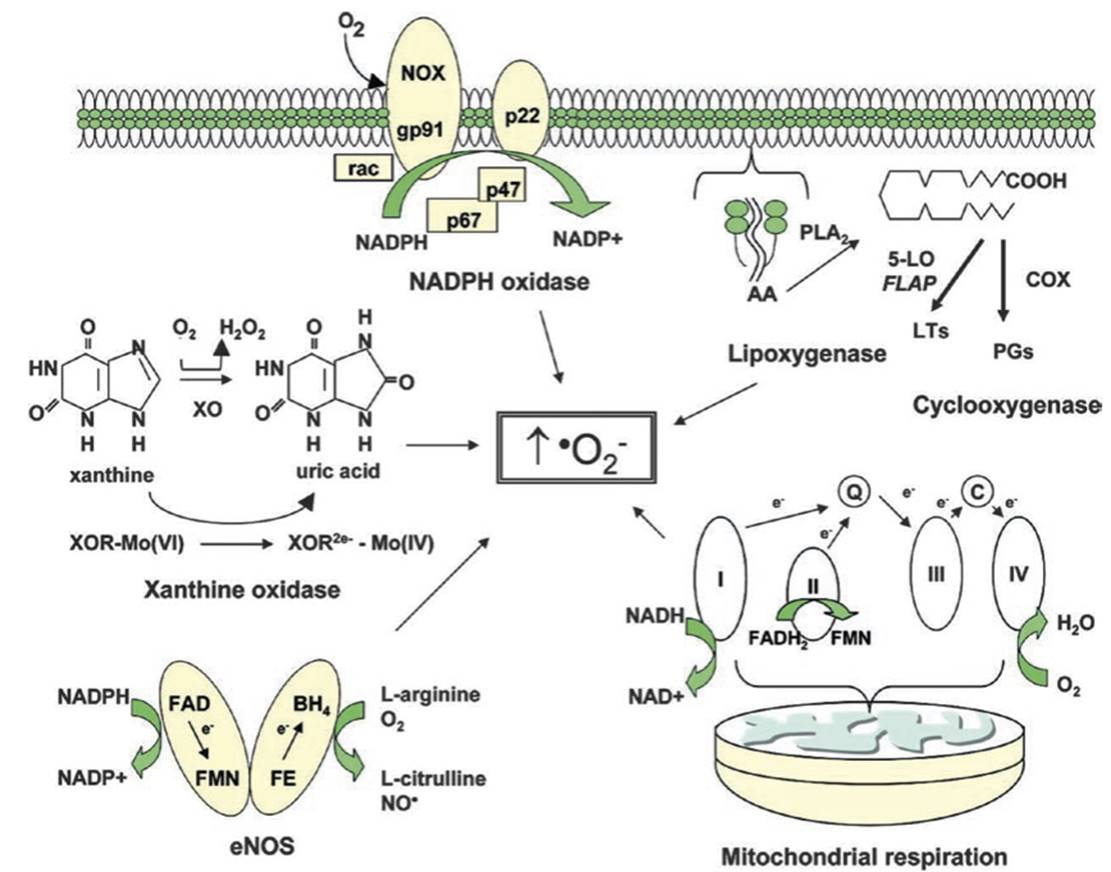
Fig. 2.3. Metabolic and enzymatic sources of superoxide anion in the vasculature. Superoxide anion (∙O2−) is formed by several metabolic and enzymatic sources within the cell. NADPH oxidase is composed of multiple membrane-bound and cytoplasmic subunits. The enzyme is activated when the cytoplasmic subunits p67 and p47 and the small G-protein Rac assemble with the membrane-bound NOX (vascular homolog of gp91phox) and p22phox. NADPH oxidase uses NADPH as a substrate and, in vascular cells, is considered an important source of reactive oxygen species (ROS) generation.
The lipoxygenases and cyclooxygenases (COX) generate ROS indirectly by promoting the formation of inflammatory mediators. Arachidonic acid (AA) that is cleaved from the cell membrane by phospholipase A2 (PLA2) is then metabolized by 5-lipoxygenase (5-LO) in the presence of its accessory protein (FLAP) to form leukotrienes (LTs). AA is also metabolized by the cyclooxygenases to form members of another family of inflammatory mediators, the prostaglandins (PGs).
Mitochondria also generate superoxide as electrons are transferred from complex I to cytochrome oxidase during normal cellular respiration. Xanthine oxidase (XO), which converts hypoxanthine and xanthine to uric acid, is an additional source of ROS. As xanthine is converted to uric acid, two electrons are donated to molybdenum (Mo) at the active site of the enzyme, thereby reducing it from Mo(VI) to Mo(IV).
Finally, endothelial nitric oxide synthase (eNOS), when substrates or cofactors are not replete, uncouples to generate superoxide in preference to NO. Abbreviations: Q coenzyme Q, C cytochrome C, NAD nicotinamide adenine dinucleotide, FAD flavin adenine dinucleotide, FMN flavin mononucleotide, FE heme iron, BH4 tetrahydrobiopterin
The ROS not only can be directly cytotoxic but also are responsible for oxidizing and peroxidizing lipid and phospholipid within LDL particles. Lipid peroxidation products (e.g., malondialde- hyde, 4-hydroxynonenal, phosphocholine of oxidized phospholipids, γ-ketoaldehydes, and 2-(ω-carboxyethyl) pyrrole) are highly reactive. For example, proteins can be rendered immunogenic when they form adducts with γ-ketoaldehydes, resulting in the activation of T cells and dendritic cells.
Atheromatous Plaque. During the initial phases of atherogenesis, macrophage foam cells that undergo programmed cell death and turn into apoptotic bodies are efficiently cleared by macrophage dependent phagocytosis. This orderly clearance process does not promote inflammation. However, as the rate of foam cell formation and accumulation increases, the milieu within the vessel wall changes. More cellular apoptosis and oncosis (ischemic death) ensues.
Phagocytic capacity is eventually exceeded, and the balance between foam cell apoptosis and clearance is lost, leading to progressive accumulation of lipid and apoptotic debris (Fig. 2.10). Fatty streaks progressively enlarge forming an atheromatous plaque which organizes with a lipid core and fibrous cap. More advanced lesions can have a necrotic core and can undergo calcification via the activity of a variety of osteogenic factors, including bone morphogenetic protein, osteonectin, and osteocalcin, among others. Plaque that is not yet fibrosed or calcified retains some degree of plasticity, as evidenced by the observation that multiple therapeutic interventions can induce plaque regression in target lesions.
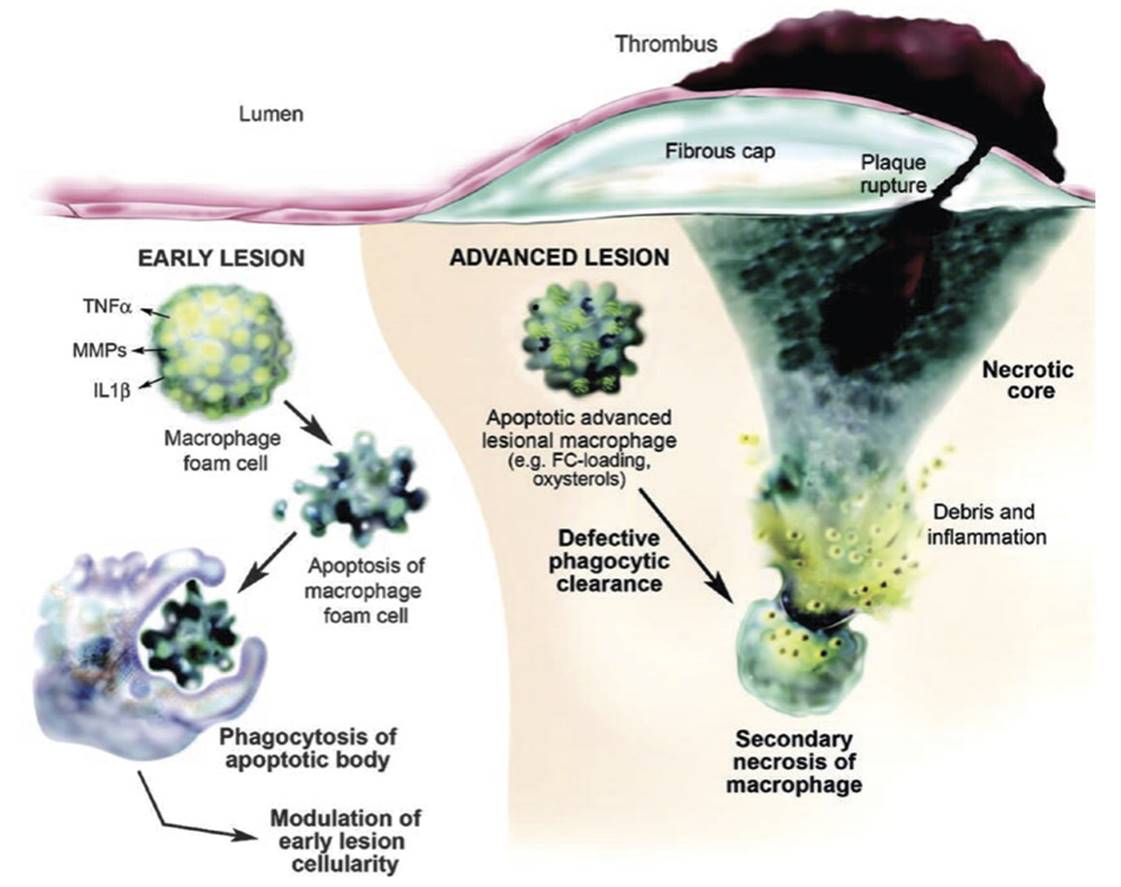
Fig. 2.10. The so-called “volcano” model of atherosclerotic plaque formation. In early atherosclerotic lesions (left), macrophage foam cells undergo apoptosis and are efficiently phagocytosed and cleared by other macrophages. This process controls lesion cellularity and rate of disease progression. However, in later lesions (right), apoptotic macrophages are not engulfed and cleared as efficiently resulting in a net accumulation of apoptotic and necrotic macrophages with the generation of a necrotic core. This leads to the mounting of an inflammatory response which can lead to plaque instability and eventual rupture
The phagocytosis of apoptotic cells and apoptotic bodies is tightly orchestrated. Apoptotic cells express a variety of “find me” (e.g., lysophosphatidylcholine, sphingosine-1 -phosphate, the fractalkine CX3CL1, and adenosine 5'-tri- phosphate and uridine-5'-triphosphate) and “eat me” (e.g., phosphatidylserine, altered ICAM-1 epitopes on the cell surface, increased calreticulin exposure) molecules that promote phagocytic cell attraction and migration, target cell discovery, and engulfment/clearance.
Apoptotic neutrophils express neutrophil-borne pentraxin-3 which promotes their recognition and removal by macrophages. Lactadherin functions as a coupling molecule that facilitates the binding of apoptotic cell phosphatidylserine to vitronectin on phagocytic macrophages. It is possible that deficiencies in these molecules may lead to impaired apoptotic cell clearance. An example of this is a deficiency in the receptor tyrosine-protein kinase MER which is associated with rapid progression and enlargement of the necrotic core in experimentally induced plaques.
As an atheromatous plaque evolves, the arterial wall reorganizes in a way that maintains luminal diameter and blood flow, a process known as positive or “Glagovian” remodeling. Plaque initially develops in an outward direction, producing vessel wall ectasia. It is only in the later stages of atheromatous plaque evolution that there develop progressive luminal obstruction and, ultimately, physiologically significant reductions in blood flow and oxygen delivery. Within the plaque, cellular necrosis promotes increased inflammation which accelerates atherogenesis and destabilizes plaques. As an illustration of just how important inflammation is in atherosclerosis, suppressing inflammation in humans with a monoclonal antibody directed against IL-1β results in a reduction of acute cardiovascular events independent of any change in serum lipoprotein levels.
Maintaining the architectural stability of a plaque is essential to preventing acute cardiovascular events. Unstable plaques typically have large lipid cores, high inflammatory tone (characterized by increased macrophage density and increased inflammatory mediator expression), and reduced smooth muscle cell density. In contrast, stable plaques are characterized by increased smooth muscle cell density, low inflammatory tone, small macrophage infiltrates, and a small lipid core. Calcification of plaque also tends to render it more stable.
Superficial surface erosions, plaque ulceration, and frank plaque rupture expose the lipid core to blood. This exposed lipids as well as tissue factors and collagen promote platelet degranulation and aggregation, resulting in the propagation of an overlying thrombus. If the thrombus completely occludes the arterial lumen, the patient experiences acute tissue ischemia. A thin fibrous cap provides less structural and tensile strength opposing plaque fracture and opening in response to a sudden stressor, such as vasospasm or hemorrhaging into the base of a plaque from injured or leaky vasa vasora. Hemorrhaging into the base of a plaque is an important cause of atheromatous plaque rupture.
A sudden rise in the volume of a plaque can lead to the loss of architectural integrity. In addition, repetitive low volume hemorrhages into the base of a plaque secondary to leaky vasa vasora can lead to cumulative trauma, increased entry of leukocytes, and increased deposition of cholesterol and other lipids in the core of the plaque. As erythrocytes are cleared from the plaque’s interior, cholesterol from cell membranes is left behind and functions as a substrate for expansion of the plaque’s lipid core (Fig. 2.11). Over time, this too can lead to plaque destabilization. The plaques that are least likely to rupture are the ones that are calcified and fibrotic.
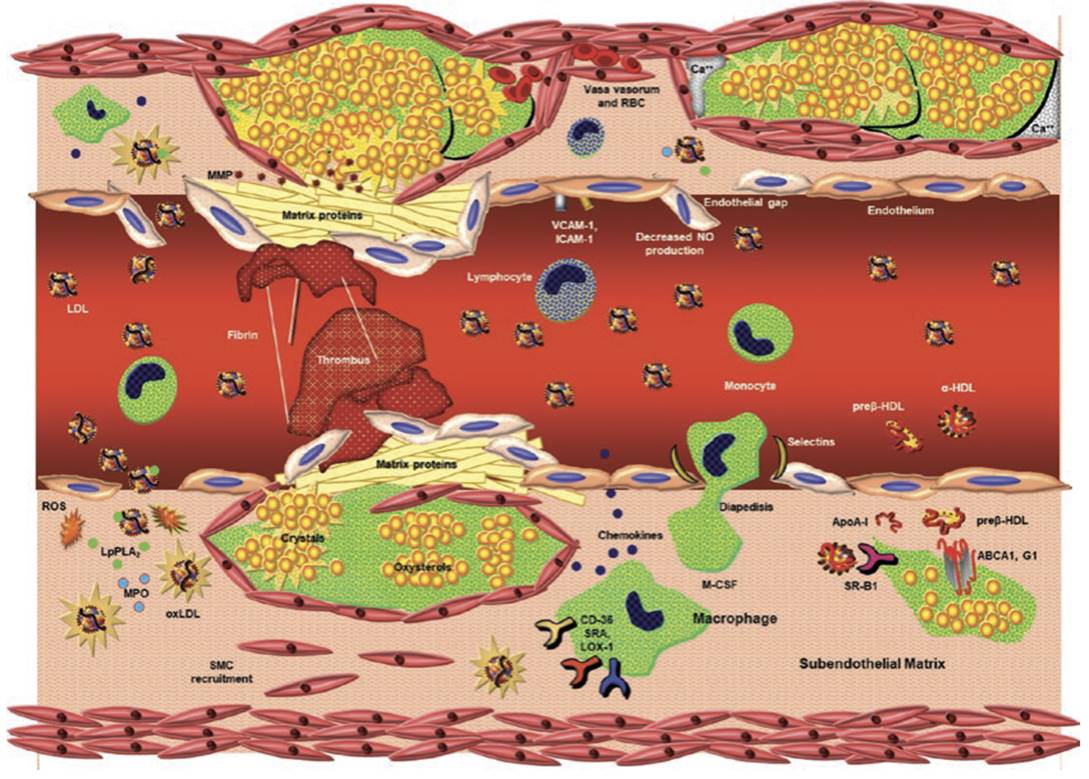
Fig. 2.11. Schematic of atherogenesis and atherosclerotic plaque rupture. Lower right quadrant. Monocyte in serum binds to selectins (ICAM-1, VCAM-1, selectin-P) on dysfunctional endothelium. The monocyte then reorganizes its actin cytoskeleton and traverses loosened gap junctions between endothelial cells in response to monocyte chemoattractant protein-1 (MCP).
Once in the subendothelial space, it can secrete interleukins and cytokines to mount an inflammatory response. As the monocyte takes up residence, it converts to a macrophage, of which there are multiple populations. By expressing such cell surface receptors as CD36, scavenger receptor A (SRA), and lectin-like oxidized low-density lipoprotein receptor (LOX-1), macrophages scavenge oxidized low-density lipoproteins (LDL) and remnant lipoproteins.
As intracellular cholesterol content increases, the macrophage becomes a progressively more lipid-enriched foam cell. In order to offload cholesterol, macrophages express the transmembrane cholesterol transport proteins ATP-binding membrane cassette transport proteins (ABC) A1 and G1, which can lipidate apoprotein A1 and spherical HDL particles, respectively. Early during atherogenesis, smooth muscle cells from the tunica media are recruited for transmigration into the intima. Upper right quadrant. Lymphocytes also bind to cell surface adhesion molecules and function as antigen presentation cells and a source of inflammatory mediators.
LDL particles enter the subendothelial space by traversing dysfunctional endothelium. Foam cells coalesce to form fatty streaks, and as lipid and cellular debris increase in volume, an atheromatous plaque forms with a lipid core. As larger amounts of cellular debris accumulate that are no longer cleared by phagocytic macrophages, a necrotic core forms. Upper left quadrant. A mature atherosclerotic plaque can be rendered unstable by bleeding into the base of the plaque via disrupted vasa vasora coursing through the tunica adventitia.
The sudden increase in blood volume at the base of the plaque raises intra-plaque pressure and can induce plaque rupture, exposing collagen and releasing adenosine 5′-diphosphate and calcium, all of which activate platelets, leading to the formation of overlying thrombus and arterial luminal occlusion. The surface of the plaque may be more prone to rupture because surface matrix proteins have been degraded by matrix metalloproteinases (MMP). Lower left quadrant. LDL particles can be oxidized by reactive oxygen species (ROS: superoxide anion, peroxynitrite, hydrogen peroxide) produced by myeloperoxidase (MPO) and a variety of lipoxygenases.
Oxidized LDL particles are scavenged by macrophages. Lipoprotein-associated phospholipase A2 (LpPLA2) hydrolyzes phospholipids into lecithin and a free fatty acid, both of which promote inflammation. Scavenged cholesterol can be stored as either pools of oxysterol or as cholesterol crystals. Cholesterol crystals can pierce through plaque surface area and promote platelet activation and thrombus formation. As atherosclerotic plaque becomes more inflamed and less stable, it can rupture, also potentiating platelet activation and thrombus formation
In the statin era, it is apparent that the percentage of ACS secondary to plaque erosion rather than acute plaque rupture has been increasing. Eroded plaques are described as having been denuded of endothelium and have increased neutrophils (and myeloperoxidase activity), decreased macrophage and T-cell constituents, small lipid cores, and large numbers of smooth muscle cells with dense proteoglycan and glycosaminoglycan intercellular matrix material.
A variety of coronary imaging studies suggest that culprit lesions giving rise to ACS have (1) large plaque volume, (2) large necrotic core, and (3) positive remodeling compared to plaques that remain stable [163]. Among patients suffering sudden death, more than 70% of ruptured plaques were characterized as having >75% luminal narrowing. In contrast, 5% of these cases were due to culprit lesions with <50% luminal narrowing. Among patients with ST-segment elevating MI, the average luminal obstruction is 66%. Typically, there is significant, rapid progression of plaque volume prior to its rupture, which can also be quite unpredictable. Identifying vulnerable plaque that will eventually rupture or fissure remains a significant unsolved issue in contemporary cardiology.
A more recently elucidated mechanism by which plaque can rupture is from the formation of cholesterol crystals within the plaque. Recent investigation shows that cholesterol can crystallize within lesions as well as perforate the plaque surface, leading to core expansion, intimal injury, and plaque instability (Fig. 2.12).
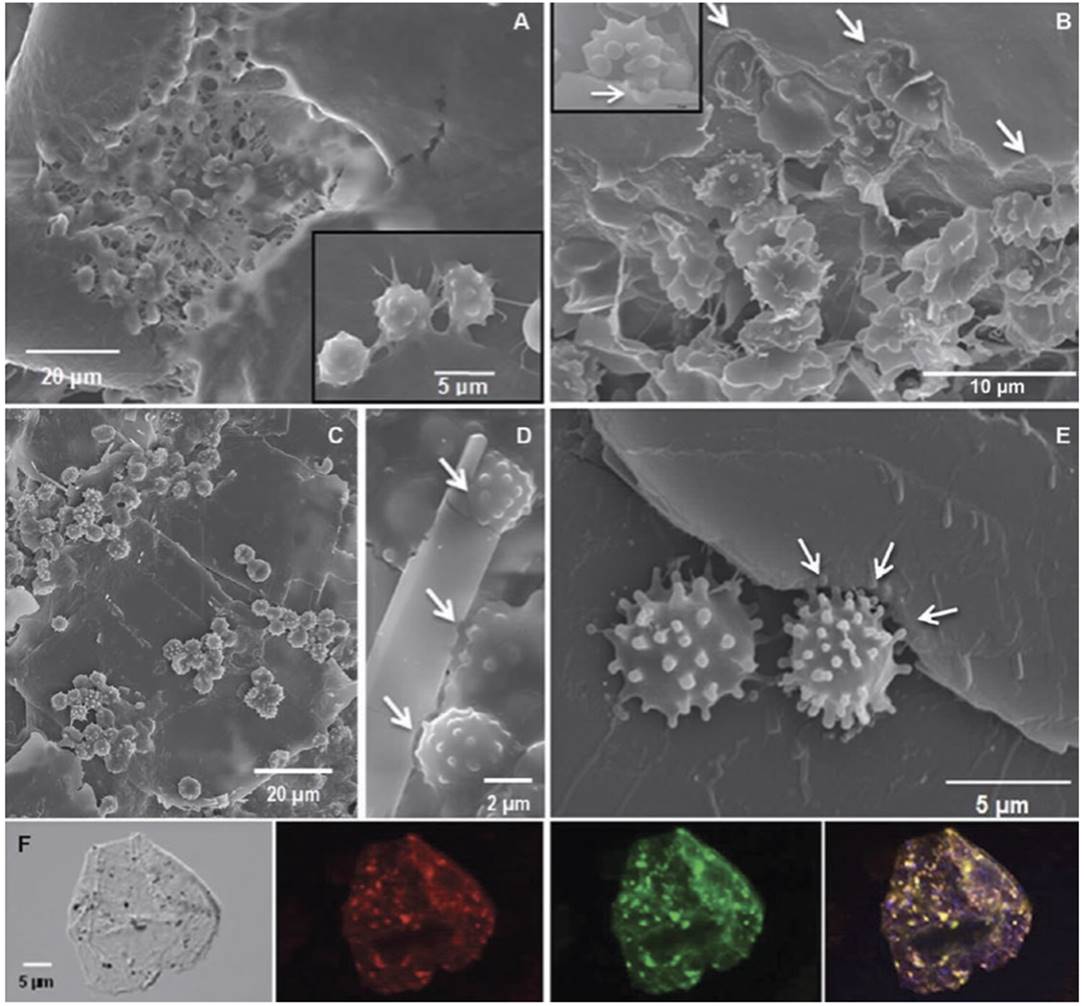
Fig. 2.12 Cholesterol crystals and atherosclerotic disease. Macrophages from coronary aspirates appear to be eroding cholesterol crystals. (a–e) Scanning electron micrographs demonstrate macrophages engaging cholesterol crystals with notched crystal matrix (arrows). Inserts demonstrate macrophage gummy attachment to the crystal edges and etching (arrow) of the crystal surface. (f) Confocal fluorescence microscopy de1monstrates cholesterol aggregates suggestive of crystalline cholesterol (yellow-green particles stained with cholesteryl Bodipy-C12) within the cytoplasm of aspirated macrophages.
The orange-red fluorescence is a specific marker for macrophages. Cholesterol deposits can be detected in the cytoplasm using differential interference contrast (shown in gray) and fluorescence microscopy (red, green, and composite image). The unstained control did not exhibit fluorescence (not shown)
Oxidized LDL scavenged by the macrophage cell surface receptor CD36 correlates with cholesterol crystallization. Cholesterol crystals augment plaque inflammation by activating nucleotide-binding domain leucine-rich repeat- containing family, pyrin domain-containing 3 inflammasome that in turn stimulates IL-1β production. Clusters of cholesterol crystals can also be released from culprit plaques during an acute MI and correlate with increased arterial narrowing and reduced reflow subsequent to percutaneous coronary intervention.
Date added: 2025-02-17; views: 508;