Life Cycle. Attachment. Penetration. Replication
Like all viruses, HIV does not have sufficient genetic material to reproduce itself. Therefore, to ensure its survival, HIV must infect a cell and redirect the cellular machinery to make viral progeny. It accomplishes this objective in a series of sequential steps that collectively define its life cycle.
HIV has four steps common to the life cycles of all viruses:
(1) attachment, during which the virus binds to the target cell;
(2) penetration, when the virus releases its genetic material and other factors needed for replication into the cell;
(3) replication, when viral genomes and proteins are made; and
(4) morphogenesis, during which new viral particles are formed and released as mature progeny (Fig. 3). While all viruses go through this cycle, each virus has evolved a unique strategy for accomplishing each step. In addition, retroviruses have steps pertaining to the reverse transcription and integration of the viral genome into the host chromosomes.
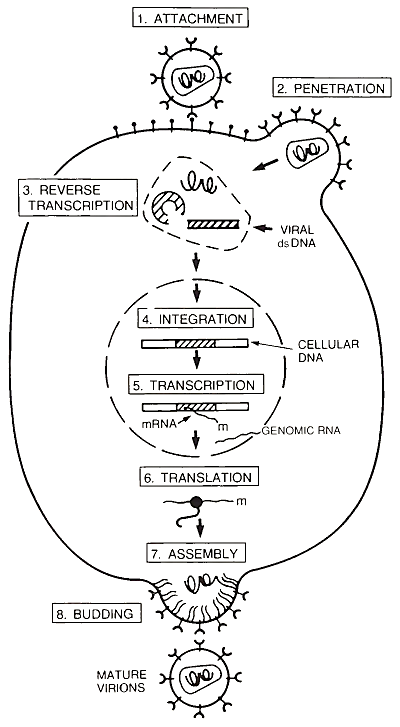
FIGURE 3. The HIV life cycle. Attachment: HIV binds to the cellular receptor. Penetration: Interaction of the HIV envelope glycoprotein and the cellular receptor leads to fusion between the viral and cellular membranes, releasing the nucleoprotein core into the cell cytoplasm. Reverse Transcription: Viral genomic RNA inside the core is copied into DNA. Integration: Viral double stranded (ds)DNA is inserted into the host cell chromosomes.
Transcription: Viral mRNA and genomic RNA are made from the viral DNA template. Translation: Viral mRNA is translated into protein. Assembly: Viral proteins and genomic RNA aggregate at the cell surface. Budding: New virions composed of the RNA genome and viral proteins are released from the cell membrane by budding. As the new virion buds, the viral protease becomes active and cleaves gag proteins, forming the nucleoprotein core
A. Attachment. HIV attaches to the target cell when its envelope glycoprotein binds to a specific molecule (i.e., receptor) on the surface of the target cell. The major receptor for HIV is the CD4 (also called T4) molecule, a surface protein that serves as a marker of the state of cellular differentiation in immuire cells. The CD4 protein is found almost exclusively on cells of the immune system, particularly on scavenger cells (e.g., monocytes and macrophages) and a subset of T lymphocytes called T helper cells.
In fact, it was depletion of the CD4+ lymphocytes seen in AIDS patients that provided the first clue that the CD4 molecule was a receptor for the virus. Subsequently, direct binding between gpl20 and the CD4 molecule has been demonstrated experimentally. Regions important to CD4-gpl20 interaction have been mapped to areas in the C-terminal half of gpl20 and to a region in the N-terminal immunoglobulin-like domain of CD4 near the OKT4A epitope.
Although cells bearing the CD4 surface antigen are the major targets for HIV (HIV is tropic for CD4-bearing cells), cells lacking surface expression of this molecule can also be infected. Clues for this finding also came from clinical observations. Many HIV-infected individuals experience neurological dysfunction not attributable to the opportunistic infections or cancers associated with AIDS. Brain and cerebrospinal fluid (CSF) specimens from such individuals frequently yield virus when placed in culture. Biological characterization of these HIV isolates from the brain has revealed that they are tropic for macrophages.
Although it is possible that the macrophages are the major producers of virus in the central nervous system, it is clear that cells of neural origin can also be infected. HIV message and protein can be found in glial cells in some infected individuals, and CD4“ glial cells can be infected by HIV in culture. Similar studies have demonstrated that some CD4“ human epithelial and fibroblast cells can also be infected.
One implication of HIV being able to infect cells that do not express CD4 is that other cellular proteins could serve as receptors for HIV. It is also not clear whether the CD4 molecule alone is a sufficient receptor. One series of experiments addressed this question by showing that, only in some cases, HIV- resistant cells lacking CD4 expression can be made susceptible to, infection by introducing the CD4 gene into the cell and causing expression of its protein on the cell surface (i.e., transfecting the cell with the CD4 gene).
Human epithelial cells successfully transfected with the CD4 gene become highly susceptible to HIV infection. However, mouse epithelial cells similarly transfected are not. In the latter case, HIV was still able to attach to the transfected mouse cells, but was unable to penetrate them. In addition, some established T cell lines with high expression of the CD4 protein do not appear to be susceptible to HIV infection. These findings suggest that a second cellular receptor involved in penetration might be required for infection, at least in some cell types.
B. Penetration. How HIV gets into cells is less well understood. In general, all enveloped viruses penetrate cells by one of two mechanisms. The first involves fusion of the virus envelope directly with the plasma membrane. The second results from internalization of the virus in the cell by receptor-mediated endocytosis, followed by fusion of the virus envelope with the membrane of the vesicle that contains it.
In the latter case the binding of the viral envelope glycoprotein with the cellular receptor triggers the cell plasma membrane to engulf the virus particle and bud off into the interior of the cell, forming a vesicle called an endosome. Once in the endosome, the virus is subjected to a decreasing pH that typically causes the envelope protein to change shape (conformation). This pH-induced change in conformation then triggers fusion and virus entry.
Several lines of evidence suggest that HIV fuses directly with the cell plasma membrane. First, in electron micrographs HIV particles can be seen fusing at the cell surface. Second, HIV entry into cells does not require a low-pH compartment (e.g., an endosome). Finally, cells that have been mutated so that the CD4 molecule cannot be internalized are still able to be productively infected. Therefore, it appears that the predominant mode of penetration into cells occurs by fusion at the cell surface, although some fusion in endocytic vesicles might also occur.
Although the steps involved in the fusion reaction are not known, the transmembrane subunit of the envelope protein (gp41) is thought to initiate the process. Presumably the 30 hydrophobic amino acids located at its amino terminus (i.e., the putative fusogenic domain; see Section I,A) directly interact with a target cell membrane, leading to fusion with the viral envelope. The hydrophobicity of this domain also suggests that it is hidden in a crevice in the interior of the envelope complex, where it is protected from the aqueous phase that surrounds the exposed part of the protein.
Therefore, if this region is active in initiating fusion, it must move into a position that enables it to interact with the target membrane. Such a change in the shape of the envelope protein complex could be an important trigger for HIV fusion with cells. This event could occur when gpl20 binds the CD4 protein or possibly when a different part of the envelope binds to another cellular receptor.
C. Replication. Once the viral core is released into the cell, the complicated process of viral replication begins. First, the viral genome (i.e., single-stranded RNA), probably still within its proteinaceous core located in the cytoplasm of the cell, is copied into a doublestranded DNA molecule by the viral enzyme reverse transcriptase. This enzyme has many complicated functions, including the ability to make DNA from a RNA template, the reverse of the usual flow of genetic information.
The molecular details of reverse transcription are not discussed here, but several features are noteworthy. Unlike the cellular RNA polymerases which stick to their DNA templates and travel down them in a continuous fashion, transcribing with high fidelity, viral reverse transcriptases skip back and forth on one template as well as between different RNA templates during a single round of transcription. Moreover, it is error prone.
While most reverse transcriptases make many more mistakes than eukaryotic polymerases, the HIV reverse transcriptase is especially imprecise; its error rate is at least 10 times higher than other viral reverse transcriptases. The clinical consequences of this characteristic are not clear, but it could allow for the spontaneous generation of new viral strains with different biological and immunological properties (see Section III,D).
The final products of reverse transcription are double-stranded DNA copies of the viral genome that have been modified so that they have approximately 630 base pair duplications flanking the ends of the protein-coding sequences. These LTRs are formed when the unique sequences at the 5' and 3' ends of the viral genome (i.e., U5 and U3) are duplicated during reverse transcription. The LTRs appear to be important for the insertion of the viral DNA into the host genome as well as for the regulation of viral gene expression. The viral enzyme integrase (i.e., endonuclease) coordinates the complicated molecular gymnastics involved in integration.
The final result of reverse transcription and integration is the establishment of the proviral state. As a provirus the viral genetic information is stably packaged in the cellular chromosomes, “indistinguishable” from the native cellular genes. Thus, viral genes are replicated along with host genes in every round of cell division, so that all progeny cells are infected. This form of passive infection (i.e., the transmission of viral genes during cell division) is independent of the production of viral progeny and occurs only in dividing cells.
However, the spread of infection often depends on the production of new viral particles by the infected cell. In order for this to happen, appropriate cellular and viral factors must be present to trigger the replication and morphogenesis pathways.
The genetic “switchboard” that regulates the amount of virus made lies in the regulatory elements in the 5' (upstream) LTR (i.e., the HIV promoter). Depending on which proteins are available to interact with these regulatory nucleotides, transcription of viral genomes and various mRNAs is turned on or off. If no viral progeny are being formed and few, if any, viral products are being made, then the provirus is said to be in a latent state. From the standpoint of the organism, the virus is silent. Alternatively, viral progeny could be produced either in small or very large amounts, usually with detrimental effects on the infected host.
As mentioned in Section I,B, the responsive element for the tat protein is encoded in the LTR. Nuclear factors from other viruses can also trans- activate the HIV LTR under experimental conditions. In general, it is believed that viral regulatory proteins exert their effects primarily through interactions with other cellular proteins. Thus, differences in HIV expression can be observed in various cell types.
The HIV LTR also has copies of regulatory elements for several other well-described cellular transcription factors, including nuclear factor кВ, (NF-kB), Spl, TATA box transcription factor (TFIID), and CCA AT transcription factor (CTF) (Fig. 4). Experimentally, many factors can bind to the HIV LTR and cause an increase in the expression of the genes under its control. These proteins could play a role in the increased HIV production seen in activated T cells. It appears that almost anything that activates T cells [e.g., foreign antigens, infectious agents, mitogens, phorbol esters, and cytokines, such as interleukin-1 (IL-1)] is associated with increased virus production in infected cells.
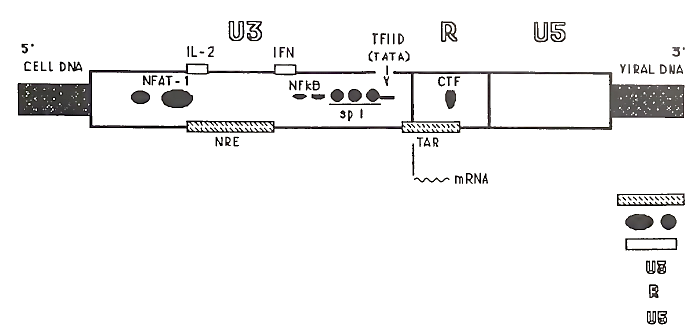
FIGURE 4. Regulatory regions in an HIV 5' long terminal repeat
Clearly, the regulation of HIV expression is no simple matter. The multitude of regulatory factors, however, strongly suggests that HIV gene expression is finely tuned. Various combinations of viral and cellular factors may allow for either very low or very high levels of virus production. Therefore, the amount of virus made in any given cell may vary enormously and may be highly responsive to physiological stimuli.
D. Morphogenesis.Soon after infection, only the mRNAs for regulatory proteins (i.e., multiply spliced transcripts) appear to be translated in detectable quantities. As mentioned in Section I,B, the rev protein plays an important role in regulating the mRNAs available for translation, but its mechanism and the cellular factors involved are not well defined. Subsequently, when the cell is committed to virus production, progeny viral genomes and mRNAs for structural proteins (i.e., long transcripts) accumulate in the cell cytoplasm.
Once in the cytoplasm, the mRNAs for structural proteins are synthesized and transported to their appropriate cellular destinations for assembly into new viral particles. The envelope glycoproteins are targeted to the cell surface, where they are anchored in the plasma membrane by the gp41 subunit. The gag and gag-pol polyprotein precursors assemble with the progeny genomes on the inner surface of the plasma membrane.
The covalently linked myristyl group at the end of the gag and gagpol precursor molecules apparently holds the molecules in position at the cell membrane, facilitating their interaction and assembly into viral cores. As the gag and gag-pol polyproteins aggregate, the cell membrane bulges outward, and the viral particle begins to take shape as it buds off from the plasma membrane. Concurrently, or shortly after the virus buds off from the cell, the viral protease becomes active, cleaving itself and the other gag and enzyme components from the gag-pol polyprotein precursor. Only then, after the viral particle is released from the cell and the mature gag and pol proteins are generated, is the new viral particle in a mature form, able to infect a new cell.
Pathogenesis. Cytopathological Effects of HIV
How HIV causes disease is the least defined aspect of HIV infection. Our understanding of the pathogenesis of AIDS is limited, because our knowledge of HIV and the immune system is incomplete and because cells from the same and different tissues communicate with each other in complex ways. What is clear, however, is that AIDS represents the final stage of years of HIV infection.
A key feature of the highly pathogenic nature of HIV almost certainly involves the virus’ preemptive attack on the immune system. Because HIV infects and kills or impairs important immune cells, it weakens that arm of the immune system. As the immune system becomes compromised, the virus may be able to replicate more, and this in turn weakens the immune system further. Such a scenario involving a vicious cycle between virus replication and immune system destruction may explain the rapid deterioration in health that commonly occurs in the later stages of HIV infection.
T Cell Depletion.The hallmark of AIDS is a depletion of T cells bearing the CD4 surface molecule (typically T helper cells) and immune dysfunction related to impairment of T-helper cell activity. T cells are critical to the proper functioning of both the cellular and humoral components of the immune system. Upon recognition of a specific antigen (i.e., a protein with distinguishing site) presented to it by an antigen- presenting cell (e.g., a macrophage), T helper cells signal other immune cells to respond to foreign antigens. This event occurs through the release of various soluble factors (i.e., cytokines) and through direct contact with other immune cells. Therefore, infected T cells can potentially affect other uninfected cells through aberrant or decreased communication.
Cytopathological Effects of HIV.Studies of lymphocytes grown in tissue culture have revealed that HIV affects CD4+ T cells in a variety of ways. The most characteristic finding is that HIV-infected T cells fuse with uninfected CD4+ T cells, forming large multinucleated giant cells (i.e., syncytia) with shortened life spans (Fig. 5). Syncytia form because the viral envelope protein (gpl20) expressed on the surface of infected cells binds the CD4 molecule of uninfected cells, inducing cell-cell fusion. Through such syncytia formation the infection can spread from cell to cell without releasing new viral particles from the cell, and all cells in the syncytia die prematurely.
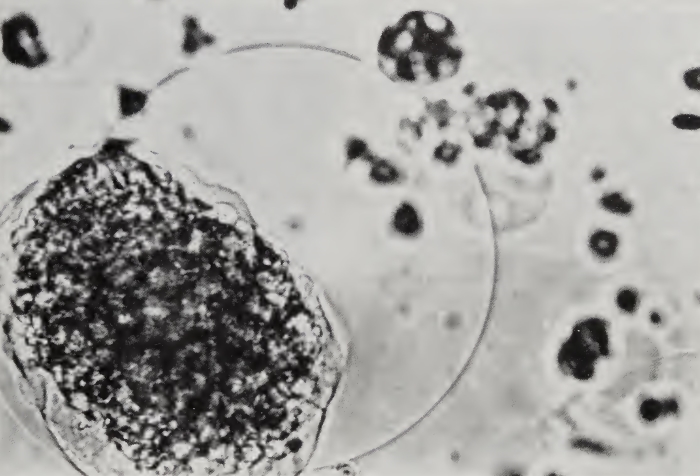
FIGURE 5. HIV-infected T cells showing syncytia and balloon degeneration. Magnification x60,000
HIV can also cause dramatic morphological changes in single cells that do not undergo fusion with other cells. Cells with such cytopathological effects (CPE) are typically enlarged and have ballooning cell membranes, signs of imminent cell death. The mechanism of such effects is not known, but several studies have provided clues to the factors involved.
One study has shown that in vitro infection of cells with a particular strain of HIV resulted in the production of extremely high levels of viral genomes and protein. Calculations from this study show that as much as 40% of the total cellular protein synthesis in these cells was involved in the production of the core gag protein.
However, it was unclear whether HIV simply outcompeted the host cell for available biosynthetic factors or whether it actively inhibited host protein synthesis. In any case, as has been shown in other viral infections (e.g., polio), extremely high levels of viral protein and genomes can be directly associated with cytocidal properties of viruses. Similarly, there are reports of large amounts of unintegrated reversed transcribed HIV genomes found in infected cells with cytopathological effects. The production and expression of large amounts of these unintegrated viral genomes could compromise host DNA and protein syntheses.
Other studies have noted changes in the properties of the membranes of infected cells. Alterations in lipid metabolism and in membrane permeability to ions have been found in infected cells. These changes could lead to rapid influxes of fluid into the infected cell, causing the cell to burst. This could explain the balloon degeneration frequently observed (Fig. 5). However, it is unclear whether such membrane perturbations are directly responsible for cytopathological effects, or are associated phenomena.
Date added: 2023-05-09; views: 960;