The diseases of metabolic exchange
Normally, genes control steps of different metabolic pathways. The gene mutation may cause decreased enzyme activity or even failure in function. There are many diseases caused by failure of one metabolic step. This group of diseases is called diseases of metabolic exchange. When enzymes can’t work at all, the metabolic precursors of reaction controlled by the enzyme are accumulated in the tissue. These accumulated substances suppress activity of surrounded cells.
This mechanism occurs in phenylketonuria, galactosemia, and alkaptonuria. On the other side, absence of the metabolite can cause a range of hereditary defects as hereditary cretinism, adrenohenital syndrome and so on. The pathology process also may occur on a level of renal tubules. The accumulated substance can be excreted improperly or not fully. According to imbalanced exchange it can be distinguished following types of metabolic exchange diseases.
Diseases of amino acid exchange. The most common example of this type is phenylalanine misbalance (pic 11.1).
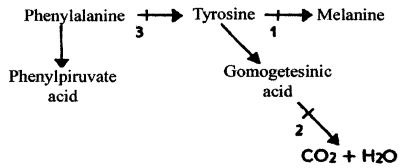
Pic. 11.1. The scheme of human phenylalanine exchange: 1-3 - the points of metabolism blocking by mutations; 1 - albinism, 2 - alkaptonuria, 3 - phenylketonuria (by M. E. Lobanov, 1967)
Phenylketonuria - is an autosomal recessive disease. It is caused by deficiency of phenylalaninhydrxylase enzyme. This enzyme converts phenylalanine to tyrosine. When it is blocked, phenylalanine is converted to phenylpyruvate and excreted with the urine. The rate of this disease in Europe is 1:10000. The signs of disease are irritability, convulsions, mental retardation, microcephaly, loss of pigmentation of skin, hair, and the iris. If newborns suffering from this disease are feed as usual newborns, they express all those signs in few months. But if we give them a diet without phenylalanine, they develop as usual children without any signs of mental retardation. To evaluate phenylketonuria, the 10% FeCl test is used. It gives green color to urine when it is positive. The express-tests are used to evaluate phenylketonuria right after delivery.
Total albinism - is an autosomal recessive disease. It is caused by a defect of the gene controlling the enzyme, which converts tyrosine to melanin. Thus, melanocytes loose their ability to produce melanin pigment. The signs of the disease are absence of melanin in skin, hair, and eyes. The eyes look of red color because of visible blood capillaries. The rate of this disease is about 1:20000.
Alkaptonuria - Is a recessive abnormality, having a rate of about 3-5:1000000. It is caused by a deficiency of homogentistic acid oxidase enzyme. Signs of the disease are special staining of cartilages and arthritis in elderly. There are diagnostic signs such as fast changing in color to dark in urine with added bases and changing in color to red with added Milon’s reactive (containing ions of Hg) which prove the presence of tyrosine in the urine.
Diseases of lipid exchange. This hereditary disease group includes familiar lipidoses characterized by excessive level of lipids in the blood and by excessive intracellular storage. The first group includes essential familiar hyperlipidemia and essential familiar hypercholesterolemia. The second group includes gangliosidoses (Tay-Sach disease), sphignomielose (Nyman-Pick disease), and cerebrosidose (Goshen disease).
The essential familiar hyperlipidemia is characterized by excessive levels of glycerids and chylomicrons and dispersed lipopropeins in blood, especially after fatty food intake. The first sign of disease is bad transparency of plasma over erythrocyte in erythrocyte sedimentation reaction. The important addition signs are xantoms, acute stomach pain with tachycardia, vomiting. These signs are also observed in patients with acute abdomen inflammation processes.
The incorrect diagnosing leads to unnecessary surgical examination. The proper therapy for such patients is a diet reduced in fat (fat consumption around 30-60g per day). The syndrome is caused by different mechanisms. Among them is suppressed chylomicron removal from blood and glycerin metabolism block. It is possible that in many cases it is inherited dominantly.
The essential familiar hypercholesterolemia is characterized by excessive levels of cholesterol and phospholipids in blood. The arthrosclerosis develop very fast. The common sign is xantoms and xantelasms on the skin and tendons. It is an incomplete dominant disease with rate 1:500. Today, one real way of treatment is an appropriate diet containing a few of cholesterol rich compounds. Instead of fat of milk and eggs, it is better to use vegetable oil.
Infantile amavrotic idioty was firstly described by E. Tay in 1881 and L. Sach in 1896. The second name of disease is Tay-Sach disease. Affected children appear normal at birth and do not usually develop the symptoms until about eight months, at which time signs of mental deterioration become evident. Within a year if birth affected children are blind; they rarely live past their fifth year. Among other signs are defects of parenchymal organs (liver, kidney), skin. Tay-Sach disease is rare in most human populations.
However, Tay-Sach disease has a high incidence among Jews of Central and Eastern Europe. Many parents were relatives, which can prove theory about local reproduction of singular mutation. Individuals homozygous for the allele lack an enzyme necessary to break down a special class of lipids called gangliosids, which occur within the lysosomes of the brain cells. As result, the lysosomes fill with gangliosids, swell, and eventually burst, releasing oxidative enzymes that kill the brain cell. There is no known cure for this condition. The rate of disease is 1:300000 in usual population.
Diseases of carbohydrate exchange. Among the diseases are diabetes mellitus, pentoseuria, fructoseuria, glycogenoses, galactosemia, and hyperbilirubinemia.
Diabetes mellitus - is an autosomal recessive disease with increased glucose blood level. The abnormal gene is wide spread (about 4-5% of homozygotes), but has a small penetrance (about 20%). Total number of patient is about 1.2-1.3% of population, whereas gluoseuria is evaluated in 2.7%.
There are two types of diabetes mellitus. The first one develops mainly in young people. It is caused by autoimmune destruction of Lanherhans insuli, which produce insulin. All cells of the body need insulin to get glucose from a blood. If there is no insulin, cells suffer from glucose deficiency, in spite of high level of glucose in blood. The one way to treat such diabetes is to eject insulin continuously throughout the life.
Diabetes with late onset is called diabetes of second type. It often occurs in obese people with arthrosclerosis. It is caused by small glucose consumption by tissues because of insulin receptors breakdown. It is treated well by sulfanilecarbamide preparations. The diabetes is diagnosed through checking blood glucose level and urine glucose level.
Diseases of steroids exchange. Main disease from this group is adrenohenital syndrome. Its rate is 1:5000 -1:67000, whereas heterozygous rate is about 1:35 - 1:128. It is an autosomal recessive disease. It is expressed in a form of hermafroditism in females and as preliminary virilization in males. Commonly it is because of hereditary hyperplasia of adrenal gland caused by inherited defects of steroid hormone biosynthesis. In the urine of such patients, we can find many androgenic 17-ketosteroids.
The natural sex of a patient is determined by evaluating sex chromatin of buccal epithelium. Clinical signs may be presented by virilization only and accompanied with adrenal failure and electrolyte imbalance. In many cases, virilization is accompanied with high blood pressure. Both males and females have early puberty development and early bone growth zone closure. The late onset is connected rather with adrenal cancer than with adrenohenital syndrome.
Diseases of purine exchange. It is gout. It is an autosomal recessive disease with incomplete penetrance (about 20%) in males and complete nonpenetrance in females. The disease develops exclusively in aged men as urate salts infiltrations in tissues. Such infiltration causes inflammatory reactions. The kidneys suffer from gout very often and kidney failure is a main cause of death of these people. Approximately 1-2% of people have hereditary asymptomatic pattem of disease, with suppressed uric acid exchange and increased level of it in an organism. During gout, uric acid concentration reach 5-16 mg%. It is provided by enhanced uric acid synthesis and decreased removing through kidneys. The gene nonpenetration in women makes genetic analysis more complicate.
Diseases of blood clotting system. They are represented by hemophilias A, B,and C. Hemophilia A - is a sex linked disease. Only men suffer from this disease. It is caused by defect of VIII coagulation factor (antihemophilic protein). Clinical sign of hemophilia is hemorrhage. The hemorrhage in hemophilia is caused by innocent reasons and it may last for hours.
The symptoms become evident in early childhood. An average life span of patient is 16-22 years. It is believed that there are 125000 patients suffering from hemophilia A in the world. In spite of each generation gene removing, the disease rate stays constant. The lost mutations are refilled by new ones. The origination of new mutations of hemophilia A occur with 1.3-4.2* 10*-5 rate. 28% of hemophilia cases are sporadic caused by new originated mutations, whereas 72% of hemophilia cases are inherited from previous generation.
Hemophilia В - is sex linked disease too. Only men suffer from this disease. It is caused by defect of IX coagulation factor. Clinical signs similar to hemophilia A. The genes, which are responsible for hemophilia A and B, are localized in different X-chromosome regions. The IX factor concentration in a patient’s blood is about 2-6% from normal value. An average life span of patient having hemophilia В is 22 years. The rate of sporadic cases is about 9%.
According to WHO data, the birth rate of hemophilia A child is 1:10000, whereas the birth rate of hemophilia В child is in 10 times less. But patients with hemophilia A die more often in early postnatal period. That’s why, hemophilia В occurs in population only in 5 times less than hemophilia A.
Hemophilia C or Willebrand disease - is an autosomal dominant disease. It is caused by rare changes in antihemophilic protein structure (factor VIII) and decreasing activity of factor preventing vessels wall damage. Patients have less ability to stop hemorrhage (women have especially long and abundant menses). Sometimes, a blood transfusion is required to treat those patients.
Defects in hemoglobin structure. Abnormal hemoglobins are evaluated mostly by electrophoresis. If hemoglobins of heterozygous individual are subjected to electrophoresis, it is revealed two different hemoglobins moving with different speed. One is normal hemoglobin A, and the second is the mutant.
The most important is hemoglobin S. Erythrocytes containing hemoglobin S become “sickled” in shape. In heterozygous individuals having Ss genotype the concentration of hemoglobin S is small, and erythrocytes express sickle shape very rare. But in homozygous individuals the hemoglobin S is abundant. Erythrocytes most of the time stay in a sickle shape and that why they are removed by spleen from blood. Sickle cell erythrocytes cause thrombosis, they are subject to massive hemolysis. That leads to homozygous death in early childhood, whereas heterozygous are clinically normal.
Heterozygous individual for T hemoglobin (dominant allele of talasemia) have no clinical signs as heterozygous individuals for hemoglobin S. But the homozygous state causes very severe erythroblastic anemia (Mediteranean anemia). Its clinical symptoms are spleen and liver enlargement, bone changes caused by compensative hyperplasia of bone marrow. Erythrocytes are produced smaller in shape with less amount of hemoglobin and they have decreased life span. If patient has talasemia, he produces hemoglobin F throughout the life.
In spite of lethal phenotype, genes S and T (and some other genes encoding defect hemoglobins C, D, E) became very spread in populations, especially in some geographic zones. It was found that gene S is wide spread among native Africans and their descendents in America; gene C - among population of Guinea Gulf; gene E - among population of South-East Asia; gene D - in West India; gene T - among population of Italia, Greece, Bengali, South-East Asia and South China. The heterozygous individuals are more resistant to Plasmodium vivax invasion.
Diseases of ions exchange. There are hepatolenticular degeneration (Wilson disease) and hemochromotosis in this group.
Wilson disease is autosome recessive disease. During this disease, ions of cuprum infiltrate liver, brain, kidney, cornea tissues. Also the excessive excretion of cuprum ions is evaluated, whereas the blood level of cuprum ions is low. The cerulloplasmine level is also small. The reabsorbtion of amino acids, glucose, uric acid and phosphate salts is failed in kidney. Pathogenesis of disease is still not clear. Half of the patients were bom in families of close relatives who were affected. Heterozygotes show decreased incorporation of Cu64 isotope to cerulloplasmine.
Hemochromatosis - is disease of ferrum storage with everyday income 24mg. It is characterized by excessive amount of hemosiderin in liver, heart, endocrine glands and tissue reaction to those infiltrations. Clinically, hemochromatosis has the following signs as liver cirrhosis, hand skin pigmentation, diabetes mellitus in men over 35 years of age.
It is very rare expressed in women. It is probably due to ferrum lost during lactation, pregnancy and menses. It is inherited dominantly with incomplete penetrance. However, some variants with early onset may have recessive pattern of inheritance. Heterozygous individuals have increased skin pigmentation, high ferrum blood level, and increased ferrum absorption from intestine.
Date added: 2023-01-09; views: 880;