Concluding Remarks: Solution-mineral Interactions in REE-bearing Hydrothermal Systems
Although the data available for the properties of REE aqueous species and REE minerals at elevated temperatures are still sparse, they permit some preliminary conclusions to be drawn about the factors controlling the behavior of these elements in hydrothermal systems. It should be noted, however, that these conclusions are still very tentative.
For example, the lack of data on the carbonate speciation of the REE in hydrothermal solutions and the limited knowledge of the properties of the main REE carbonate phases preclude reliable evaluation of the behavior of the REE in carbonate-bearing systems. Moreover, considering that effectively all the experimental data available on the behavior of the REE in hydrothermal solutions have been collected at acidic conditions, it is likely that reliability of any models based on these data will be less for alkaline and weakly alkaline, than for acidic and weakly acidic conditions.
Additional uncertainties may also be brought to our conclusions by the fact that the models for REE behavior described above are based on the properties of pure end-member minerals and do not account for their ability to form solid solutions due to the lack of information on the properties of the latter. However, most of the naturally occurring REE minerals are solid solutions, and the reactivity of their components can potentially be significantly different from that of the corresponding pure end-members, potentially shifting or even altering the trends and relationships discussed here.
Bearing in mind the above caveat, we conclude that at elevated temperatures the main transporting ligands for the REEs are chloride and sulfate. In contrast to hydroxyl and fluoride, these two ligands form solids with the REEs that are relatively soluble, and, therefore, the concentrations of the REEs that can be developed in chloride- and sulfate-bearing solutions are not restricted by saturation with solids (for the range of REE concentrations encountered in natural systems).
Although REE chloride complexes are only weakly stable, this effect is outweighed by the very high concentrations of chloride typical of natural hydrothermal solutions. Figure 8 compares predominance fields for Nd aqueous species at 25 and 300 °C as a function of the activity of chloride and sulfate in aqueous solutions. From this figure, it can be seen that the contribution of simple hydrated REE3+ ions (Nd3+ in this case) to the mass balance of dissolved REEs decreases significantly with temperature.
Immobilization of the REEs in aqueous solutions due to the precipitation of REE-bearing solids occurs when REE-rich fluids encounter phosphate- or fluoride-rich environments; the highly insoluble minerals mon- azite/xenotime (REEPO4) or fluocerite (REEF3) form, respectively. It is also likely that interaction with a F/CO3-enriched environment will lead to the deposition of highly insoluble REE fluorocarbonate minerals, e.g. bastnäsite-(Ce) and parisite-(Ce). How this would occur, however, is still unclear due to the lack of information on the behavior of the REEs in CO3-bearing hydrothermal systems.
In some natural systems, there is evidence that hydrothermal processes have led to the fractionation of the heavy REEs from the light REEs. For example, in the Ranger unconformity-type uranium deposit (Australia), the La/Lu ratio increases by almost an order of magnitude from low values proximal to the ores to high values distal from them.
In principle, this can be explained by the fact that, although the chemical properties of the REEs are very similar at ambient temperature, at hydrothermal conditions they change systematically with atomic number of the lanthanides. Indeed, as shown earlier (Figure 2), the formation constant, ß 1, for REE chloride complexes decreases by over an order of magnitude from La to Lu at 250 °C.
Thus, La is predicted to be considerably more mobile than Lu, potentially explaining the observed behavior of these elements in the example cited above. A similar effect has been observed for REE fluoride complexes. However, as discussed above, it is unlikely that fluoride complexes contribute significantly to the hydrothermal mobility of the REEs due to the very low solubility of REE fluoride solids.
Another potential cause of REE fractionation is the differential solubility of REE minerals. Thus, minerals with lower solubility will saturate in solutions earlier than those with higher solubility and be deposited before the latter. If deposition occurs across physicochemical gradients that are spatially distributed, and if the minerals that deposit differ in their proportions of the REEs, their differential solubility will lead to fractionation of the REEs.
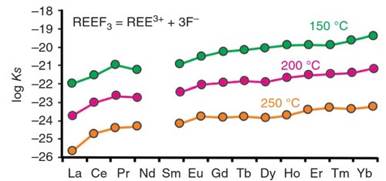
Figure 9. The distribution of values for the solubility product of REE fluorides at 150, 200 and 250 °C. Source: Migdisov et al. 2016. Reproduced with permission of Elsevier
For example, the solubility product of fluocerite varies considerably with atomic number, increasing by two to three orders of magnitude from Ce to Lu, depending on temperature (Figure 9), and thus, its deposition could lead to fractionation of the REEs.
Finally, fractionation of the REEs can occur during interaction of REE-bearing solutions with a nominally REE-free host rock or hydrothermally formed minerals. Incorporation of the REEs in the crystallographic structure of these minerals is a complex process controlled by the ionic radii of the incorporated REEs, their charges, and the speciation of the REE in aqueous solutions.
Date added: 2023-10-03; views: 869;