Mutation: The Molecular Engine of Evolutionary Diversity and Genomic Innovation
Mutation.Mutations are the ultimate source of most variation in all organisms. By definition (e.g., Barton et al. 2007), they consist of changes in the genetic message (either at the level of the gene or the chromosome) that are heritable and, implicitly, detectable. Mutation generates the variation; recombination amplifies this by mixing it, in essence by repackaging it, not by creating brand new packages. We note in passing that molecular biologists frequently speak of inherent mutation rates (usually referring to replacement rates of one DNA base by another, as may be read, for example, from a genomic sequence), whether or not there is a change in the wild-type phenotype.
This fundamental level of mutation may go undetected if the protein is not functionally altered, if two or more mutations compensate for each other giving a pseudo wild-type, or if another gene product takes over the function of the altered protein. Thus, there are really three mutation rates—the inherent rate of uncorrected change in DNA sequences; the rate at which the mutants survive; and the rate at which survivors are detected in a population by virtue of their phenotypic differences. Although there are many types of mutation, fundamentally most involve errors introduced into the genome.
These may occur spontaneously or be induced by endogenous or exogenous mutagens, typically occurring during replication; and errors in recombination, repair, or in large-scale chromosomal rearrangements (Alberts et al. 2015; Griffiths et al. 2015). To these long-established sources of failure in cellular machinery must now be added the more recently recognized category of genomic change caused by mobile genetic elements such as transposons (below).
Mutations are mechanically inevitable—damage to DNA occurs and copying, proofreading, and segregating mechanisms are not and cannot be perfect. For this reason Williams (1966, p. 12) has argued that mutations are not adaptive, i.e., they should not be considered to be a means for ensuring evolutionary plasticity. That they still occur is largely in spite of natural selection rather than because of it (Williams 1966, p. 139); However, this does not mean that there cannot be selection for a particular mutation rate and there are clear differences in rates among species (below). Overall, because many of these alterations are deleterious, there has been strong selection pressure (reduced reproductive fitness) over millennia to reduce rates in organisms. Incremental benefit in increased fidelity below a certain point may be offset by greater physiological costs in correction (Kimura 1967; Drake 1991). It has also been argued (Lynch 2010) that the lower limit is set by genetic drift.
Regardless, there is some value in having background genetic ‘noise’ (Drake 1974; Drake et al. 1998). This is illustrated by the essential function of mutation (somatic hypermutation) in antibody genes of the B lymphocyte cells, which contributes to antibody diversity in vertebrates (French et al. 1989). Analogous genetic rearrangements provide the operon structure needed for expression of nif genes in a nitrogen-fixing cyanobacterium, Anabaena (Golden et al. 1985). The terminal step in heterocyst differentiation involves excision of DNA in response to an environmentally triggered, site-specific DNA recombinase (Haselkorn et al. 1987). Thus, much the same rearrangement process is used in a prokaryote and an advanced eukaryote.
With the mechanistic characterization of transposable elements in both prokaryotes and eukaryotes (for an overview, see Griffiths et al. 2015), the distinction at the molecular level between mutation and recombination is often one of semantics. Classification schemes are more-or-less subjective because the degree of genetic change occurs as a continuum. For instance, where relatively large segments of DNA (hundreds or thousands of nucleotides) are rearranged, such as by inversion or exchange between chromosomes, the event is often called mutation by molecular biologists (Chaps. 9 and 11 in Watson et al. 2008). Yet it falls within the domain of recombination (in the sense of mixing of nucleotides) as defined by eukaryote geneticists. For example, site-specific recombination (see Recombination, later) is a mechanism that can lead to mutation.
Probably the best-known form of recombination is the even exchange of sequences between aligned, homologous chromosomes at meiosis. However, recombination can occur between any two regions of sufficient DNA similarity on the same chromosome or between paired chromosomes, an event known as ectopic (nonallelic) recombination. This potentially leads to mutation due, for example, to deletions or inversions (in the case of intrachromosomal recombination) or, if two chromosomes are involved, to unequal crossing-over where one offspring receives too many copies of the sequence and the other too few (Chap. 12, Barton et al. 2007).
Before proceeding into the details, some important general points about mutations need to be made. First, they occur in somatic as well as in germline cells, the implications of which will be discussed in Sect. 2.5 (see Shendure and Akey 2015). Suffice it to say here that, with few exceptions (such as among the somatic cell lines that produce antibodies), high rates must be avoided in both lineages. The implications of mutation in somatic cells tend to be overlooked because of the focus by evolutionary biologists on the germline.
Second, the impact of a mutation will depend on the environment, broadly construed (Chap. 7). A mutation deleterious in one set of circumstances could be beneficial in another. Also, as discussed later, while mutation rates can vary with environmental conditions, the environment does not induce mutations that are specifically adaptive; i.e., adaptively directed mutation does not occur (Barton et al. 2007; Futuyma 2009). Current evolutionary thinking is that mutation and selection are separate processes.
It was well established for bacteria by Luria and Debruck in the 1940s, confirmed by the Lederbergs in the 1950s, and reconfirmed with elaborations since then, that mutations conferring resistance to phage or antibiotics happen both before and after the selective agent is applied and are random with respect to their adaptive value (Barton et al. 2007). (For commentary on the controversy that some mutations in bacteria are adaptive, allegedly being ‘directed’ by the environment, see Sniegowski and Lenski 1995; Foster 2000; Sniegowski 2005.)
Third, the impact of a mutation can depend on the stage of development of an organism. Mutations occurring in early ontogeny, for example, in animals at the blastula stage, are much more likely to be lethal because of their far-reaching impact on subsequent differentiation. In contrast, those occurring later may affect relatively superficial properties such as eye color (Bonner 1965, pp. 123-128; 1988, p. 168). As Bonner notes, it follows from this that any mutation at any stage in the life of a unicellular organism is more likely to be lethal than would a similar mutation during most of the life of a macroorganism.
Extent of mutations If we categorize mutations by the degree of the base changes involved, the first kind consists of simple base position (typically called point) substitutions such as a C to a T in the nucleotide sequence, or by addition or deletion of a single base (Graur and Li 2000; Griffiths et al. 2015). Because of the degeneracy feature of the genetic code (‘wobble’ in the third codon position), often there is no change in the amino acid sequence (‘synonymous’ or ‘silent’ mutation). Mutations may also be silent if a related amino acid is inserted (missense conservative mutation) or if the affected region of the gene product is unimportant. If the altered codon specifies a different amino acid, the change is a missense nonconservative mutation.
This is manifested either as an altered but functional (‘leaky’) protein product or as a defective protein. The latter category is very common, as in sickle cell anemia, Tay-Sachs disease, and phenylketonuria. One form of retinitis pigmentosa is apparently caused by a C to A transversion in codon 23, corresponding to a proline to histidine substitution (Dryja et al. 1990). Finally, if translation is terminated (nonsense mutation), a shortened and likely defective protein results.
It should be emphasized that point and other small changes in the noncoding regions of a gene, such as in regulatory sequences, can potentially be very significant in evolutionary terms. For example, such changes may create or interrupt a binding site thereby changing or even obliterating the expression of the gene. In other words, alteration in gene regulation, accomplished in multiple ways, has arguably had an even greater impact than actual mutation in the polypeptide coding regions of those same genes (see later section and Chap. 7; also Doebley and Lukens 1998; Carroll et al. 2005).
Second in extent of nucleotides involved are small insertions or deletion (indel) mutations involving one or a few base pairs. When this happens within regions coding for polypeptides, the mutations are called frameshift if the reading frame of the codons is shifted out of phase (Barton et al. 2007; Alberts et al. 2015). This can lead to premature termination or obliteration of a stop codon, so a frameshift mutation may result in a truncated, possibly unstable protein, or it may generate a completely nonfunctional product, depending on its position in the structural gene. In general, the foregoing changes, except deletions, are revertible to wild-type. Reversion of deletions depends on the nature of the deletion and the encoded gene product.
The third category of mutation comprises the potentially large-scale chromosomal changes such as bigger deletions, inversions, duplications, and insertions (transpositions) that intergrade with recombination, discussed later. Thus, the size or number of genes in chromosomes may be increased or decreased; genes may change in location (by inversion or translocation); and there may be wholesale changes in the number of chromosomes. Where a deletion is large enough to remove or disrupt a gene coding for a critical enzyme, the result is likely to be death of the cell or organism. At the other extreme, while polyploidy (more than two chromosome sets) is generally considered to be an abnormal condition, it appears to be well tolerated by some organisms, especially plants.
Here, it is estimated to have resulted in 7% of the speciation events in ferns and 2-4% in angiosperms (Otto and Whitton 2000), as well as in the associated evolution of plant gene families and structural complexity (De Bodt et al. 2005). Most such polyploidizations are followed by gene deletions over time resulting in eventual diploidy. Plants, however, again are distinctive in being able to tolerate imperfectly matched chromosomes that result from loss or rearrangement (Walbot and Cullis 1983). Stanley (1979) has emphasized the role of chromosomal rearrangement together with gene regulation in ‘quantum speciation’.
Gene expansion, contraction, duplication, or loss may contribute significantly to the evolution of complexity, functional diversity, and adaptation to the environment over evolutionary or contemporary time (see Chap. 4 and also: Verstrepen et al. 2005; Hittinger and Carroll 2007; Pranting and Andersson 2011; Andersson et al. 2015). A classic example is the evolution of the globin gene family in multicellular animals and how duplication was instrumental in creating new proteins (Hoffmann et al. 2012; Alberts et al. 2015). Analogously, the evolutionary expansion of enzyme families in plants associated with specialized metabolism for which plants are famous is attributable to various kinds of gene duplication events (Moore and Purugganan 2005). In contrast, genes can be inactivated by insertion of mobile DNA elements into the chromosome and subsequently eroded or lost.
These insertions may be fragments (generally 1,000-2,000 nucleotides) that do not code for any characters beyond those needed for their own transposition (simple transposon or insertion sequence; discussed later in Recombination section). Alternatively, they may be longer elements (complex transpo- son or transposable element) that code for a product such as antibiotic resistance in bacteria. The rates of gene loss and gain are particularly dynamic in prokaryotes due in large part to a process of horizontal or lateral gene transfer (discussed later under Sex and Adaptive Evolution in Prokaryotes), offset by inactivation and deletion (Mira et al. 2001; Lerat et al. 2005). Genome plasticity is also notable in plant (Ibarra-Laclette et al. 2013) and fungal (Raffaele and Kamoun 2012) evolution.
Rates As alluded to above, mutation rates can be estimated by recording spontaneous phenotypic changes over time in a population of organisms in nature (animals or plants) or in the laboratory (tissue culture or microbial culture). Because the frequencies of most mutations are very low in natural populations and can only be detected in large sample sizes, it is common to examine specific proteins electrophoretically or DNA sequences directly (e.g., in restriction fragment length polymorphisms, or as single nucleotide variants on a genomewide basis). The mutations tallied and mutational rates published typically pertain to single nucleotide substitutions and, in some cases, indels or copy number variants (e.g., Lynch 2010; Shendure and Akey 2015), but rarely thus far on large structural (>20 bp) variants (Campbell and Eichler 2013; Kloosterman et al. 2015).
Rates are expressed in numerous ways, e.g., on the basis of per base pair per cell replication; per genome per replication; or per genome per sexual generation (unless otherwise stated, typically on a haploid basis). For an example of such data see Table 2.3 and Lynch (2010). Different eukaryotes (or even sexes within a species) have different numbers of cell divisions involved in gamete production per sexual generation. In humans there are about 30 germline cell divisions in females, but more than 100 for spermatogenesis in males (Crow 2000; Barton et al. 2007), which thus have a higher mutation rate on a generational basis. Higher eukaryotes have a genome up to several orders of magnitude larger than viruses or prokaryotes, but most of this is in introns and inter-genic regions where most mutations are neutral rather in than functional genes.
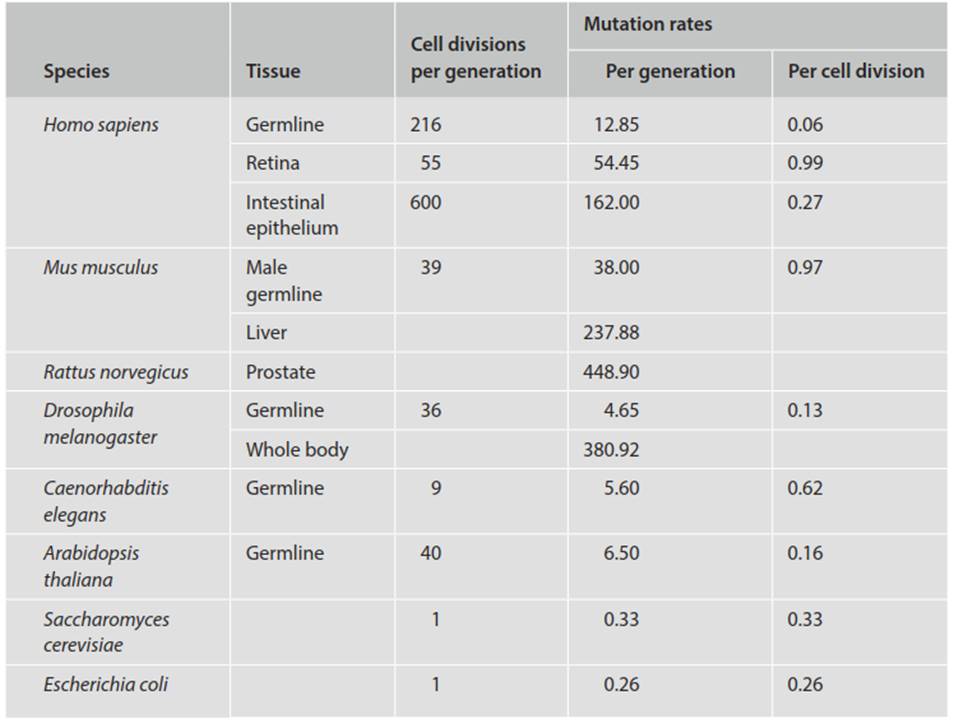
Table 2.3. Mutation rates per nucleotide site (×10-9) for selected species and tissues (abbreviated from Lynch 2010)
Thus, rates for macrorganisms are often expressed as per effective genome (i.e., excluding the genomic regions where most mutations are neutral) per replication (cell division) or per sexual generation (Drake et al. 1998). There are further complications or caveats with eukaryotes, among them age- or sex-related effects alluded to above, as well as substantially higher rates in somatic versus germline cells, discussed later. Nevertheless, as a generality, when expressed as number of mutations per base pair per replication, there are appreciably higher rates for smaller than larger genomes.
When expressed on a per genome basis, there is less variation across genome sizes or species. Rates in microbes are typically about 1/300 per genome per replication (versus about 1 per genome per replication event in the lytic RNA viruses; Drake et al. 1998). For the higher macroorganisms, on an effective genome basis, rates per sexual generation are in the range of 0.1-100, but on a cell division basis per effective genome are approximately comparable to the 1/300 for microorganisms (Drake et al. 1998; Hershberg 2015). The remarkable comparability across taxa remains to this day a source of animated debate as to, for example, whether it reflects an inherent rate of change or a common level of error correction. The implication of various determinants on the ultimate mutation rate, including factors such as sexual versus asexual reproduction and population size, has been discussed at length (see e.g., Drake et al. 1998; Lynch 2010; Sung et al. 2012).
Beyond the differences noted above, the incidence of mutation in a local sense varies with numerous other factors. Among these are the specific genomic region (so-called ‘hotspots’ vs. ‘coldspots,’ in part affected by degree of DNA superhelicity; see Foster et al. 2013), organelle (nucleus vs. mitochondrion), and type of base (AT-biased rather than GC). Thus, whereas mutation is generally described as a random phenomenon, where it occurs in the genome is far from random. The consequences of mutation are inextricably linked to life cycle (Chap. 6). For instance, mutations are masked by diploidy and even more so by polyploidy. In relatively simple organisms (such as algae with a prominent haploid gameto- phyte phase, see 7Chap. 4) the individual experiences selection mostly in the haploid stage of the life cycle. Under such circumstances mutations will not be masked and there will be strong selection pressure against mutant cells. Possibly this furthered the maintenance of haploidy, as well as the correlation of diploidy with more complex development (Otto and Orive 1995; this point is also developed in Chap. 6). In tetraploids like the agronomic crop alfalfa, mutations are hidden, but selfing results in inbreeding depression. This may be due to loss of third-order interactions or homozygous deleterious recessive alleles (Jones and Bingham 1995).
The molecular clock Notwithstanding the substantial variation in mutation patterns among species, it was recognized several decades ago that the average rates of amino acid or nucleotide substitution are approximately constant for a particular protein or gene among taxa (Kimura 1987). This fairly steady accumulation in sequence divergence over time has been called the ‘molecular clock’ (Zuckerandl and Pauling 1965; Wilson et al. 1977; Kumar 2005). Presumably, it reflects the fact that the same functional constraints exist for a given gene or gene product in different organisms. Clock analysis involves comparing the amino acid or gene sequences in a particular highly conserved protein (such as hemoglobin), or base sequences in a given gene, in several species. For protein comparisons, each discrepancy is assumed to represent a stable change in a codon corresponding to the altered amino acid. In this analysis, the original (ancestral) state, being extinct, is of course unavailable for comparison. So the versions of the sequence that appear in two or more living relatives of the phyla of interest are compared and expressed as units of accumulated amino acid or nucleotide changes.
The organisms are then arranged in a branching diagram drawn to minimize the number of changes needed to describe all the permutations from the ancestral sequence. The number of stable genetic changes can then be related to chronological estimates of evolutionary time, obtained from fossil records, since the species diverged from the common ancestor (recall Chap. 1). At a detailed level, molecular clocks have proven in general to give more resolution and reliability than the fossil record (cf. Chap. 1).
How and why the rates have stabilized where they have is unknown. An inference is often made that the clock is centrally standardized, ticking away uniformly for all species, much like a radioactive decay process, but this is not strictly the case (Jukes 1987). Substitutions are nonlinear such that irregularities occur and these may or may not reconcile over time. Also, for species of both microorganisms and macroorganisms, the clock can tick at different rates among genes, even when only silent substitution rates are considered (Sharp et al. 1989).
Nevertheless, among other interesting comparative observations are the following by Ochman and Wilson (1987): (i) the silent mutation rate for protein-coding genes in Salmonella typhimurium and Escherichia coli is comparable to that in the nuclear genes of invertebrates, mammals, and flowering plants; (ii) the average substitution rate for 16S rRNA of bacteria is similar to that of 18S rRNA in vertebrates and flowering plants; (iii) the rate for 5S rRNA is about the same for bacteria and eukaryotes.
To summarize, the present indications are that the rates of DNA divergence for a gene encoding a given function are approximately the same regardless of the organism. From this generality we derive not only very useful phylogenetic information but also insight into the origin of sequence variations and how gene families diverge (Kumar 2005).
Finally, it should be noted that the rate of phylogenetic change evidently is not controlled by the mutation rate. Rather, the evolutionary history of the major groups of organisms appears to depend on ecological opportunities afforded the simple or complex genetic variants (Wright 1978, pp. 491-511). Evolution, overall, has been for increased size and complexity, although there are several exceptions (Chap. 4). Evolutionary rates, unlike mutation rates, vary greatly even along single phyletic lines. For some existing genera (e.g., the lungfishes and the opossum among animals; horsetails [Equisetum] and club mosses [Lycopodium] among plants), evolution has been virtually at a standstill for hundreds of millions of years (Wright op. cit.). Where genetic variation has provided for a major and entirely new way of life, swift adaptive radiation follows.
Examples include the development of feathers, wings, and temperature regulation in the case of the birds; efficient limbs, hair, temperature regulation, and mammary glands in the mammals. Among prokaryotes, genes can be transmitted horizontally and in blocks among distant relatives with major evolutionary implications; see later discussion with respect to bacteria in Sect. 2.3. Other ecological opportunities are presented when new forms colonize an area where either the niches are unoccupied or are better filled by the mutant than the wild-type (Wright 1978).
Date added: 2025-06-15; views: 387;